TadA-Bench: A Million-Variant Benchmark for Future-Round Discovery Toward Agentic Protein Engineering
Pith reviewed 2026-06-28 19:59 UTC · model grok-4.3
The pith
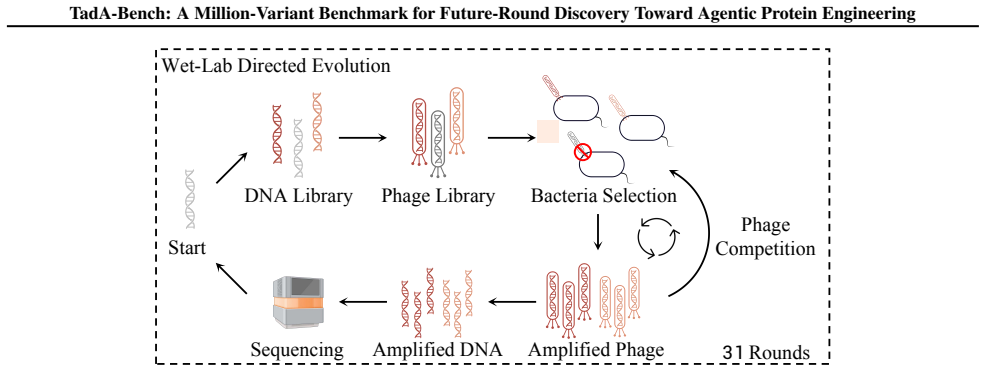
TadA-Bench requires models to rank protein variants that only appear in later directed-evolution rounds from earlier data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
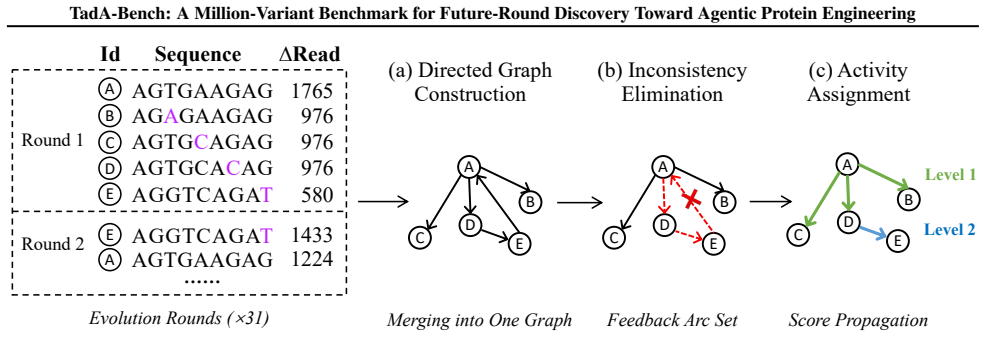
TadA-Bench preserves campaign chronology and defines a fixed-data replay task in which models, given earlier experimental rounds, must rank variants that appear only in later rounds; Seq2Graph reconciles noisy enrichment measurements into consistent cross-round activity labels, and controlled tests show that evolutionary coverage supplies more information than local data density for this future-round prediction.
What carries the argument
Seq2Graph, a graph-based label-unification pipeline that converts noisy enrichment measurements from multiple rounds into consistent activity labels across the entire campaign.
If this is right
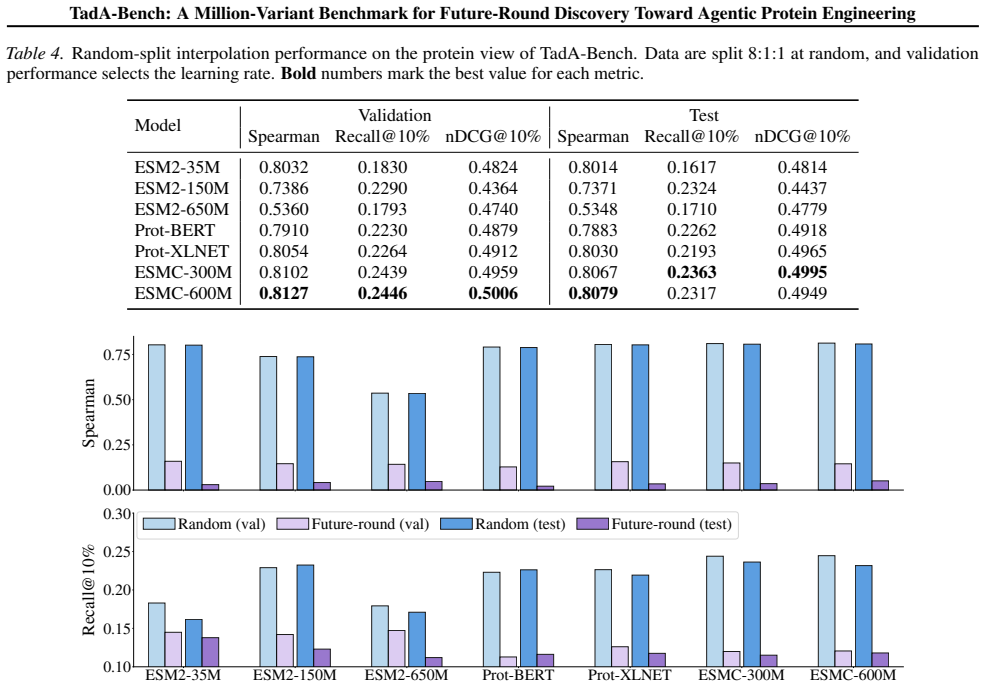
- Random data splits that ignore campaign order overestimate how well models will perform on real sequential experiments.
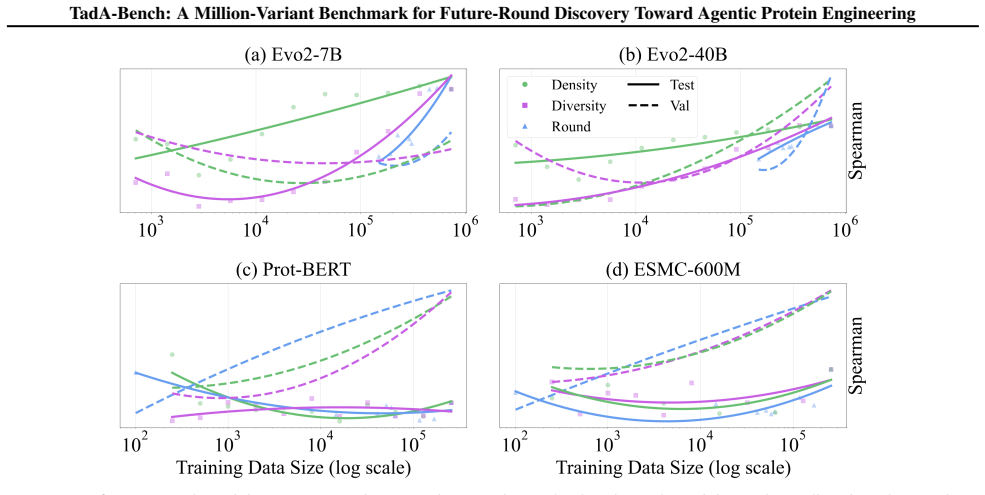
- Covering the evolutionary trajectory across rounds yields better prediction of later variants than concentrating measurements in local sequence neighborhoods.
- Even when models can rank future variants, selecting a small budgeted set of candidates for the next round remains difficult.
- The benchmark supplies a reproducible substrate that can be used to develop and compare methods for choosing the next wet-lab experiments.
Where Pith is reading between the lines
- Agentic systems trained under this replay setting would need explicit mechanisms for tracking which regions of sequence space have already been explored in prior rounds.
- The same chronology-preserving structure could be applied to other directed-evolution campaigns to test whether the coverage-versus-density pattern generalizes.
- Models might improve on the benchmark by learning to propose sequences that both extend the evolutionary path and maintain measurable activity.
Load-bearing premise
The Seq2Graph pipeline produces unbiased and consistent activity labels from the original noisy enrichment measurements.
What would settle it
An experiment in which a model trained only on the first k rounds achieves future-round ranking accuracy comparable to its random-split accuracy on the full dataset would show that the claimed separation between interpolation and future-round prediction does not hold.
Figures




read the original abstract
AI for scientific discovery is entering an agentic era, where protein-engineering systems are expected to prioritize future wet-lab experiments rather than merely fit static measurements. We introduce TadA-Bench, a million-variant wet-lab replay benchmark from 31 TadA directed-evolution rounds for future-round discovery toward agentic protein engineering. TadA-Bench preserves the campaign chronology and defines a fixed-data replay task: given earlier experimental rounds, models rank variants that appear only in later rounds. It provides aligned DNA, RNA, and protein views, and uses Seq2Graph, a graph-based label-unification pipeline, to reconcile noisy enrichment measurements into consistent cross-round activity labels. Random-split controls show strong interpolation, but future-round ranking and finite-budget candidate selection are much weaker. Controlled analyses suggest that evolutionary coverage is more informative than local data density, positioning TadA-Bench as a reproducible wet-lab replay substrate for future-round discovery toward agentic protein engineering; the data and code are released on Hugging Face and GitHub.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces TadA-Bench, a million-variant benchmark derived from 31 chronological rounds of TadA directed-evolution experiments. It defines a fixed-data replay task in which models trained on earlier rounds must rank variants that appear only in later rounds, using the Seq2Graph graph-based pipeline to unify noisy per-round enrichment measurements into consistent cross-round activity labels. Aligned DNA/RNA/protein sequences are provided. Random-split interpolation succeeds while future-round ranking and finite-budget selection are substantially weaker; controlled analyses indicate that evolutionary coverage outperforms local data density. The dataset and code are released on Hugging Face and GitHub.
Significance. If the unified labels are shown to be free of round-dependent bias, TadA-Bench would supply a valuable, reproducible wet-lab replay substrate for evaluating agentic protein-engineering systems on genuine chronological extrapolation rather than static interpolation. The emphasis on evolutionary coverage versus local density offers a concrete, testable hypothesis for future method development.
major comments (2)
- [Abstract] Abstract (paragraph on Seq2Graph): the central claim that Seq2Graph produces 'consistent cross-round activity labels' from noisy enrichment measurements is load-bearing for all future-round ranking results, yet the manuscript provides no quantitative validation (intra-variant label variance across rounds, correlation with orthogonal assays, or sensitivity to graph-construction hyperparameters). Without such checks the reported gap between random-split and chronological performance cannot be confidently attributed to biology rather than label-construction artifacts.
- [Abstract] Abstract (future-round ranking paragraph): the conclusion that 'evolutionary coverage is more informative than local data density' rests on controlled analyses whose construction details (how coverage and density are operationalized, how round index is controlled) are not specified; if these metrics are themselves derived from the Seq2Graph labels, the comparison risks circularity.
Simulated Author's Rebuttal
We thank the referee for the careful reading and the specific concerns raised about validation of the Seq2Graph pipeline and the construction of the controlled analyses. We address each point below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract] Abstract (paragraph on Seq2Graph): the central claim that Seq2Graph produces 'consistent cross-round activity labels' from noisy enrichment measurements is load-bearing for all future-round ranking results, yet the manuscript provides no quantitative validation (intra-variant label variance across rounds, correlation with orthogonal assays, or sensitivity to graph-construction hyperparameters). Without such checks the reported gap between random-split and chronological performance cannot be confidently attributed to biology rather than label-construction artifacts.
Authors: We agree that quantitative validation of label consistency is needed to support attribution of the performance gap. In revision we will add (i) intra-variant label variance computed for all variants appearing in multiple rounds and (ii) sensitivity analysis across graph-construction hyperparameters (edge thresholds, aggregation functions). The TadA dataset contains no orthogonal assay measurements, so correlation with independent activity readouts cannot be provided; we will explicitly note this limitation and its implications for the benchmark. revision: partial
-
Referee: [Abstract] Abstract (future-round ranking paragraph): the conclusion that 'evolutionary coverage is more informative than local data density' rests on controlled analyses whose construction details (how coverage and density are operationalized, how round index is controlled) are not specified; if these metrics are themselves derived from the Seq2Graph labels, the comparison risks circularity.
Authors: We will expand the Methods and Results sections to specify the exact operationalizations: evolutionary coverage is defined via average phylogenetic distance to sequences observed in prior rounds plus round-participation counts; local data density is defined via minimum edit distance to the training-set sequences. Round index is treated as an explicit blocking variable independent of activity labels. Both metrics are computed solely from sequence identity and round metadata; they do not use the Seq2Graph-derived activity values, eliminating circularity. The corresponding analysis code will be added to the public repository. revision: yes
- Correlation of Seq2Graph labels with orthogonal wet-lab assays, because no such measurements exist in the TadA directed-evolution dataset.
Circularity Check
No significant circularity; benchmark dataset release is self-contained
full rationale
The paper's core output is a released million-variant dataset and fixed-data replay task definition drawn from existing wet-lab TadA evolution rounds. Seq2Graph is presented as a graph-based processing pipeline to unify noisy enrichment scores into cross-round labels, but the manuscript does not derive any quantitative prediction, fitted parameter, or uniqueness result that reduces to its own inputs by construction. No equations equate a claimed output to a fitted input, no self-citation chain bears the central claim, and the evolutionary-coverage analysis is an empirical observation on the released data rather than a tautological renaming. The work is therefore self-contained against external benchmarks and receives the default non-circularity finding.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Seq2Graph pipeline reconciles noisy enrichment measurements into consistent cross-round activity labels without systematic bias
Reference graph
Works this paper leans on
-
[1]
Proteins: Structure, Function, and Bioinformatics , volume=
A large-scale experiment to assess protein structure prediction methods , author=. Proteins: Structure, Function, and Bioinformatics , volume=. 1995 , publisher=
1995
-
[2]
Proceedings of the National Academy of Sciences , volume=
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences , author=. Proceedings of the National Academy of Sciences , volume=. 2021 , publisher=
2021
-
[3]
Google AI , volume=
Welcome to the era of experience , author=. Google AI , volume=
-
[4]
Journal of Chemical Information and Modeling , year=
All that glitters is not gold: Importance of rigorous evaluation of proteochemometric models , author=. Journal of Chemical Information and Modeling , year=
-
[5]
Journal of chemical information and modeling , volume=
Large-scale modeling of sparse protein kinase activity data , author=. Journal of chemical information and modeling , volume=. 2023 , publisher=
2023
-
[6]
Journal of chemical information and modeling , volume=
Comparative assessment of scoring functions: the CASF-2016 update , author=. Journal of chemical information and modeling , volume=. 2018 , publisher=
2016
-
[7]
Science , volume=
Rapid in silico directed evolution by a protein language model with EVOLVEpro , author=. Science , volume=. 2024 , publisher=
2024
-
[8]
2020 , url =
Moult, John and Fidelis, Krzysztof and Kryshtafovych, Andriy and Schwede, Torsten and Topf, Maya , title =. 2020 , url =
2020
-
[9]
The Power of Scale for Parameter-Efficient Prompt Tuning
The power of scale for parameter-efficient prompt tuning , author=. arXiv preprint arXiv:2104.08691 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[10]
arXiv preprint arXiv:2110.07602 , year=
P-tuning v2: Prompt tuning can be comparable to fine-tuning universally across scales and tasks , author=. arXiv preprint arXiv:2110.07602 , year=
-
[11]
International Conference on Medical Image Computing and Computer-Assisted Intervention , pages=
Text-guided foundation model adaptation for pathological image classification , author=. International Conference on Medical Image Computing and Computer-Assisted Intervention , pages=. 2023 , organization=
2023
-
[12]
2009 IEEE conference on computer vision and pattern recognition , pages=
Imagenet: A large-scale hierarchical image database , author=. 2009 IEEE conference on computer vision and pattern recognition , pages=. 2009 , organization=
2009
-
[13]
International journal of computer vision , volume=
Imagenet large scale visual recognition challenge , author=. International journal of computer vision , volume=. 2015 , publisher=
2015
-
[14]
Advances in neural information processing systems , volume=
Imagenet classification with deep convolutional neural networks , author=. Advances in neural information processing systems , volume=
-
[15]
Proceedings of the IEEE conference on computer vision and pattern recognition , pages=
Deep residual learning for image recognition , author=. Proceedings of the IEEE conference on computer vision and pattern recognition , pages=
-
[16]
An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale
An image is worth 16x16 words: Transformers for image recognition at scale , author=. arXiv preprint arXiv:2010.11929 , year=
work page internal anchor Pith review Pith/arXiv arXiv 2010
-
[17]
Nature protocols , volume=
Phage-assisted continuous and non-continuous evolution , author=. Nature protocols , volume=. 2020 , publisher=
2020
-
[18]
Nature Communications , volume=
Phage-assisted evolution of highly active cytosine base editors with enhanced selectivity and minimal sequence context preference , author=. Nature Communications , volume=. 2024 , publisher=
2024
-
[19]
Nature , volume=
Accurate multiplex gene synthesis from programmable DNA microchips , author=. Nature , volume=. 2004 , publisher=
2004
-
[20]
Genome biology , volume=
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 , author=. Genome biology , volume=. 2014 , publisher=
2014
-
[21]
Genome biology , volume=
A scaling normalization method for differential expression analysis of RNA-seq data , author=. Genome biology , volume=. 2010 , publisher=
2010
-
[22]
Theory in biosciences , volume=
Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples , author=. Theory in biosciences , volume=. 2012 , publisher=
2012
-
[23]
Bioinformatics , volume=
DeCoDe: degenerate codon design for complete protein-coding DNA libraries , author=. Bioinformatics , volume=. 2020 , publisher=
2020
-
[24]
Protein Science , volume=
GGAssembler: Precise and economical design and synthesis of combinatorial mutation libraries , author=. Protein Science , volume=. 2024 , publisher=
2024
-
[25]
PLoS neglected tropical diseases , volume=
Degenerate sequence-based CRISPR diagnostic for Crimean--Congo hemorrhagic fever virus , author=. PLoS neglected tropical diseases , volume=. 2022 , publisher=
2022
-
[26]
The Plant Journal , volume=
Utilizing codon degeneracy in the design of donor DNA for CRISPR/Cas9-mediated gene editing to streamline the screening process for single amino acid mutations , author=. The Plant Journal , volume=. 2024 , publisher=
2024
-
[27]
nature , volume=
Highly accurate protein structure prediction with AlphaFold , author=. nature , volume=. 2021 , publisher=
2021
-
[28]
Science , volume=
Accurate prediction of protein structures and interactions using a three-track neural network , author=. Science , volume=. 2021 , publisher=
2021
-
[29]
science , volume=
A programmable dual-RNA--guided DNA endonuclease in adaptive bacterial immunity , author=. science , volume=. 2012 , publisher=
2012
-
[30]
Nature protocols , volume=
Genome engineering using the CRISPR-Cas9 system , author=. Nature protocols , volume=. 2013 , publisher=
2013
-
[31]
Nature Communications , volume=
Machine learning-guided co-optimization of fitness and diversity facilitates combinatorial library design in enzyme engineering , author=. Nature Communications , volume=. 2024 , publisher=
2024
-
[32]
Nature , pages=
Accurate structure prediction of biomolecular interactions with AlphaFold 3 , author=. Nature , pages=. 2024 , publisher=
2024
-
[33]
Science , volume=
Generalized biomolecular modeling and design with RoseTTAFold All-Atom , author=. Science , volume=. 2024 , publisher=
2024
-
[34]
Nature Portfolio , year=
Upgraded adenine base editor (uABE) with minimized RNA off-targeting activity , author=. Nature Portfolio , year=
-
[35]
Molecular Therapy , volume=
A precise and efficient adenine base editor , author=. Molecular Therapy , volume=. 2022 , publisher=
2022
-
[36]
Engineered base editors with reduced bystander editing through directed evolution , author=. Nature Biotechnology , year=. doi:10.1038/s41587-025-02937-w , publisher=
-
[37]
Cell Research , pages=
Zero-shot prediction of mutation effects with multimodal deep representation learning guides protein engineering , author=. Cell Research , pages=. 2024 , publisher=
2024
-
[38]
Cell , volume=
An AsCas12f-based compact genome-editing tool derived by deep mutational scanning and structural analysis , author=. Cell , volume=. 2023 , publisher=
2023
-
[39]
Plant Physiology , volume=
High-throughput methods for genome editing: the more the better , author=. Plant Physiology , volume=. 2022 , publisher=
2022
-
[40]
Natural Product Reports , year=
Accelerating enzyme discovery and engineering with high-throughput screening , author=. Natural Product Reports , year=
-
[41]
Frontiers in Genetics , volume=
Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes , author=. Frontiers in Genetics , volume=. 2023 , publisher=
2023
-
[42]
Nucleic Acids Research , volume=
Expanding the flexibility of base editing for high-throughput genetic screens in bacteria , author=. Nucleic Acids Research , volume=. 2024 , publisher=
2024
-
[43]
Nature biotechnology , volume=
GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases , author=. Nature biotechnology , volume=. 2015 , publisher=
2015
-
[44]
Genome biology , volume=
DeepCRISPR: optimized CRISPR guide RNA design by deep learning , author=. Genome biology , volume=. 2018 , publisher=
2018
-
[45]
Nature biotechnology , volume=
Large language models generate functional protein sequences across diverse families , author=. Nature biotechnology , volume=. 2023 , publisher=
2023
-
[46]
bioRxiv , pages=
Crowdsourced Protein Design: Lessons From the Adaptyv EGFR Binder Competition , author=. bioRxiv , pages=. 2025 , publisher=
2025
-
[47]
Bioinformatics , volume=
Generalizable sgRNA design for improved CRISPR/Cas9 editing efficiency , author=. Bioinformatics , volume=. 2020 , publisher=
2020
-
[48]
Nucleic Acids Research , volume=
Engineered Cas9 variants bypass Keap1-mediated degradation in human cells and enhance epigenome editing efficiency , author=. Nucleic Acids Research , volume=. 2024 , publisher=
2024
-
[49]
Scaling Learning Algorithms Towards
Bengio, Yoshua and LeCun, Yann , booktitle =. Scaling Learning Algorithms Towards
-
[50]
and Osindero, Simon and Teh, Yee Whye , journal =
Hinton, Geoffrey E. and Osindero, Simon and Teh, Yee Whye , journal =. A Fast Learning Algorithm for Deep Belief Nets , volume =
-
[51]
2016 , publisher=
Deep learning , author=. 2016 , publisher=
2016
-
[52]
arXiv preprint arXiv:2406.05540 , year=
A Fine-tuning Dataset and Benchmark for Large Language Models for Protein Understanding , author=. arXiv preprint arXiv:2406.05540 , year=
-
[53]
arXiv preprint arXiv:2402.13418 , year=
EvolMPNN: Predicting Mutational Effect on Homologous Proteins by Evolution Encoding , author=. arXiv preprint arXiv:2402.13418 , year=
-
[54]
Nature communications , volume=
Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning , author=. Nature communications , volume=. 2021 , publisher=
2021
-
[55]
Nature biotechnology , volume=
Large dataset enables prediction of repair after CRISPR--Cas9 editing in primary T cells , author=. Nature biotechnology , volume=. 2019 , publisher=
2019
-
[56]
Proceedings of the 24th international conference on Machine learning , pages=
Learning to rank: from pairwise approach to listwise approach , author=. Proceedings of the 24th international conference on Machine learning , pages=
-
[57]
Proceedings of the 23rd annual international ACM SIGIR conference on Research and development in information retrieval , pages=
IR evaluation methods for retrieving highly relevant documents , author=. Proceedings of the 23rd annual international ACM SIGIR conference on Research and development in information retrieval , pages=
-
[58]
Addison Wesley google schola , volume=
Modern information retrieval , author=. Addison Wesley google schola , volume=
-
[59]
Proceedings of the 42nd international ACM SIGIR conference on research and development in information retrieval , pages=
CEDR: Contextualized embeddings for document ranking , author=. Proceedings of the 42nd international ACM SIGIR conference on research and development in information retrieval , pages=
-
[60]
Proceedings of the 42nd international ACM SIGIR conference on research and development in information retrieval , pages=
Deeper text understanding for IR with contextual neural language modeling , author=. Proceedings of the 42nd international ACM SIGIR conference on research and development in information retrieval , pages=
-
[61]
Proceedings of the 44th International ACM SIGIR Conference on Research and Development in Information Retrieval , pages=
B-PROP: bootstrapped pre-training with representative words prediction for ad-hoc retrieval , author=. Proceedings of the 44th International ACM SIGIR Conference on Research and Development in Information Retrieval , pages=
-
[62]
Science , volume=
Sequence modeling and design from molecular to genome scale with Evo , author=. Science , volume=. 2024 , doi=
2024
-
[63]
2025 , eprint=
Nature Language Model: Deciphering the Language of Nature for Scientific Discovery , author=. 2025 , eprint=
2025
-
[64]
Science , volume=
Evolutionary-scale prediction of atomic-level protein structure with a language model , author=. Science , volume=. 2023 , doi=
2023
-
[65]
bioRxiv , pages=
LucaOne: Generalized Biological Foundation Model with Unified Nucleic Acid and Protein Language , author=. bioRxiv , pages=. 2024 , publisher=
2024
-
[66]
Communications Biology , volume=
A foundational large language model for edible plant genomes , author=. Communications Biology , volume=. 2024 , publisher=
2024
-
[67]
Nature , volume=
Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage , author=. Nature , volume=. 2016 , publisher=
2016
-
[68]
Nature , volume=
Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage , author=. Nature , volume=. 2017 , publisher=
2017
-
[69]
IEEE Transactions on Pattern Analysis and Machine Intelligence , volume=
ProtTrans: towards cracking the language of life’s code through self-supervised learning , author=. IEEE Transactions on Pattern Analysis and Machine Intelligence , volume=
-
[70]
bioRxiv , pages=
Saprot: Protein language modeling with structure-aware vocabulary , author=. bioRxiv , pages=. 2023 , publisher=
2023
-
[71]
Nature medicine , volume=
Therapeutic genome editing: prospects and challenges , author=. Nature medicine , volume=. 2015 , publisher=
2015
-
[72]
Genome research , volume=
Enabling functional genomics with genome engineering , author=. Genome research , volume=. 2015 , publisher=
2015
-
[73]
Nature biotechnology , volume=
Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity , author=. Nature biotechnology , volume=. 2020 , publisher=
2020
-
[74]
bioRxiv , pages=
Design of highly functional genome editors by modeling the universe of CRISPR-Cas sequences , author=. bioRxiv , pages=. 2024 , publisher=
2024
-
[75]
Nature , volume=
Illuminating protein space with a programmable generative model , author=. Nature , volume=. 2023 , publisher=
2023
-
[76]
Nature , volume=
De novo design of protein structure and function with RFdiffusion , author=. Nature , volume=. 2023 , publisher=
2023
-
[77]
Nature biotechnology , volume=
Sparks of function by de novo protein design , author=. Nature biotechnology , volume=. 2024 , publisher=
2024
-
[78]
Nature biotechnology , volume=
Machine learning for functional protein design , author=. Nature biotechnology , volume=. 2024 , publisher=
2024
-
[79]
Proteins: Structure, Function, and Bioinformatics , volume=
Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12 , author=. Proteins: Structure, Function, and Bioinformatics , volume=. 2018 , publisher=
2018
-
[80]
Chemical Science , volume=
PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences , author=. Chemical Science , volume=. 2024 , publisher=
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.