SpliceBind: Isoform-Aware Prediction of Binding Pocket Druggability
Pith reviewed 2026-06-28 12:03 UTC · model grok-4.3
The pith
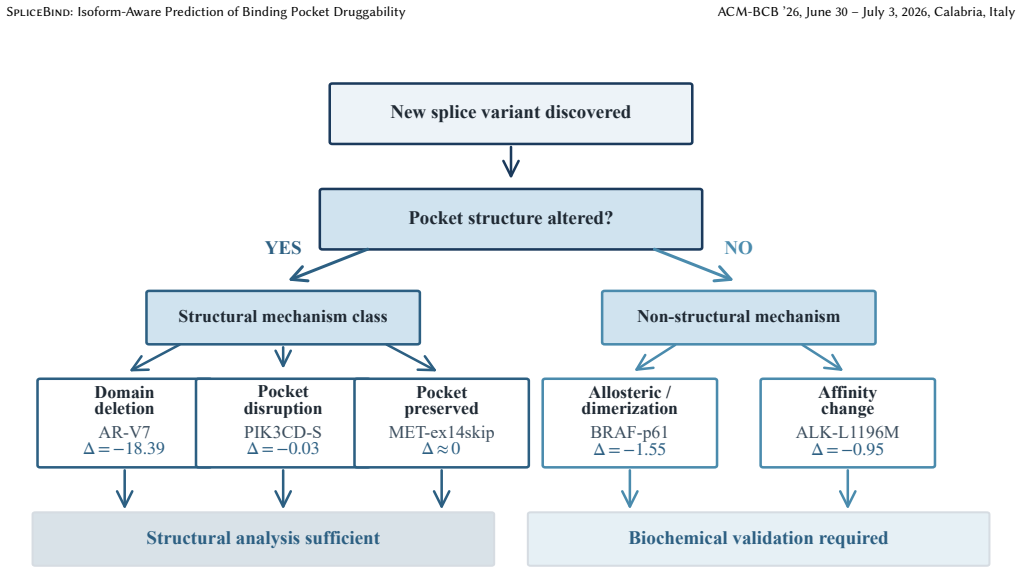
Splice-mediated resistance divides into two tiers: some mechanisms alter binding pockets detectably while others remain invisible to any pocket-based method.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
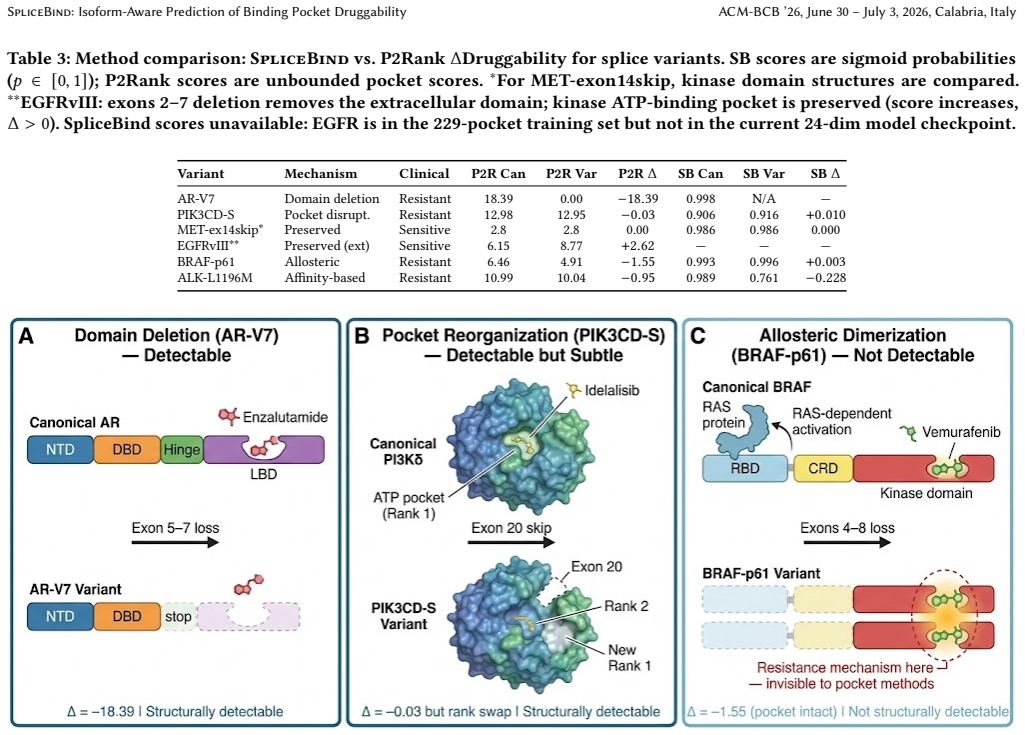
Systematic analysis of six clinically validated variants spanning five mechanism classes reveals a two-tier resistance taxonomy. Domain deletions such as AR-V7 and pocket disruptions produce structurally detectable changes, while allosteric mechanisms such as BRAF-p61 remain fundamentally invisible to any pocket-centric approach. SpliceBind reaches AUROC 0.703 versus 0.634 for P2Rank on 229 kinase pockets and its embeddings capture some affinity-based signals missed by geometry alone, as in ALK-L1196M.
What carries the argument
The two-tier resistance taxonomy that classifies splice variants according to whether they generate detectable changes in binding pocket druggability scores.
If this is right
- Clinicians can classify a newly discovered splice variant immediately to decide whether computational triage is sufficient or biochemical validation is required.
- Domain deletion variants produce large negative changes in predicted druggability that structural methods can identify.
- Allosteric variants cannot be distinguished by any refinement of pocket-centric algorithms.
- The model maintains performance on held-out kinase families at AUROC 0.761.
- Learned embeddings detect some non-geometric resistance signals that pure geometry tools miss.
Where Pith is reading between the lines
- Separate non-pocket methods such as dynamics-based or allosteric-site predictors would be needed to address the invisible tier.
- The same structural-versus-nonstructural split could be tested on other mutation classes beyond splicing.
- Clinical pipelines could route variants through the taxonomy first to reduce unnecessary lab testing.
Load-bearing premise
The six clinically validated variants represent the full range of splice-mediated resistance mechanisms and the network embeddings capture affinity effects beyond geometry in a generalizable way.
What would settle it
Discovery of an allosteric splice variant where any pocket prediction method correctly flags a drop in druggability, or a domain-deletion variant where no pocket method detects the expected change.
Figures


read the original abstract
Splice-mediated drug resistance occurs in up to 40% of patients on targeted kinase inhibitors, yet state-of-the-art druggability tools operate on single structures and cannot compare across isoforms. We introduce SpliceBind, a graph neural network framework for isoform-aware druggability prediction. Beyond improving prediction accuracy (AUROC 0.703 vs. P2Rank 0.634, p = 0.026), we address a more fundamental question: when do structural methods succeed, and when must they fail? Systematic analysis of six clinically validated variants spanning five mechanism classes reveals a two-tier resistance taxonomy. Domain deletions (AR-V7, Delta = -18.39) and pocket disruptions produce structurally detectable changes, while allosteric mechanisms (BRAF-p61) remain fundamentally invisible to any pocket-centric approach -- a boundary no algorithmic improvement can cross. Notably, learned embeddings capture affinity-based resistance missed by geometry alone (ALK-L1196M: Delta_SB = -0.228 vs. Delta_P2Rank = -0.95), partially bridging the structural-biochemical gap. On 229 kinase pockets spanning 25 families, SpliceBind achieves AUROC 0.703 (p = 0.026 vs. P2Rank) with robust generalization to held-out families (AUROC 0.761). This taxonomy transforms clinical workflows: upon discovering a splice variant, clinicians can immediately determine whether computational triage suffices or biochemical validation is required -- reducing time from variant discovery to therapeutic decision.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SpliceBind, a graph neural network framework for isoform-aware prediction of binding pocket druggability. It reports an AUROC improvement to 0.703 versus P2Rank's 0.634 (p=0.026) on a dataset of 229 kinase pockets spanning 25 families, with robust held-out family generalization (AUROC 0.761). Beyond the predictive tool, the central contribution is a two-tier resistance taxonomy derived from systematic analysis of six clinically validated splice variants across five mechanism classes, asserting that domain deletions and pocket disruptions yield structurally detectable changes while allosteric mechanisms (e.g., BRAF-p61) are fundamentally invisible to any pocket-centric method, with learned embeddings partially capturing affinity effects missed by geometry (e.g., ALK-L1196M).
Significance. If substantiated, the work offers both a practical improvement in isoform-aware druggability prediction with held-out generalization and a taxonomy that could streamline clinical triage by identifying when structural computation suffices versus when biochemical validation is required. The explicit attempt to delineate intrinsic boundaries of pocket-centric methods, rather than solely reporting accuracy gains, represents a valuable framing if the supporting evidence is strengthened.
major comments (3)
- [Abstract and Results (resistance taxonomy)] Abstract and Results section on resistance taxonomy: The claim that allosteric mechanisms 'remain fundamentally invisible to any pocket-centric approach -- a boundary no algorithmic improvement can cross' rests on systematic analysis of only six variants with a single allosteric exemplar (BRAF-p61). This N=6 sample cannot distinguish model-specific or feature-specific failure from an intrinsic limitation applying to every pocket-centric method.
- [Results (ALK-L1196M analysis)] Results (ALK-L1196M case): The statement that learned embeddings capture affinity-based resistance missed by geometry (Delta_SB = -0.228 versus Delta_P2Rank = -0.95) is presented without controls demonstrating that the effect is not an artifact of the training distribution or specific to the GNN architecture, separate from the reported held-out family generalization.
- [Methods (dataset and evaluation)] Methods (dataset construction and evaluation protocol): The manuscript reports AUROC 0.703 (p=0.026) and held-out AUROC 0.761 on 229 pockets across 25 families, but lacks explicit description of family selection criteria, splice-variant mapping procedure, and safeguards against data leakage that would be required to confirm the generalization claim is load-bearing for the taxonomy.
minor comments (2)
- [Abstract] The statistical test underlying p=0.026 is not named, nor is any correction for multiple comparisons mentioned.
- [Results (variant table/figure)] Table or figure presenting the six variants should explicitly list mechanism class, Delta values, and structural detectability classification for each to support the taxonomy.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback on our manuscript. We address each major comment below, with revisions indicated where we agree changes are needed.
read point-by-point responses
-
Referee: [Abstract and Results (resistance taxonomy)] Abstract and Results section on resistance taxonomy: The claim that allosteric mechanisms 'remain fundamentally invisible to any pocket-centric approach -- a boundary no algorithmic improvement can cross' rests on systematic analysis of only six variants with a single allosteric exemplar (BRAF-p61). This N=6 sample cannot distinguish model-specific or feature-specific failure from an intrinsic limitation applying to every pocket-centric method.
Authors: We acknowledge that the sample of six variants, including only one allosteric exemplar, is small and cannot conclusively prove an intrinsic limitation for all possible pocket-centric methods. The taxonomy is derived from the mechanistic classes in these clinically validated cases, where BRAF-p61 shows no direct pocket alteration. We will revise the abstract and Results to qualify the statement as applying to the analyzed mechanism classes and variants, emphasizing that allosteric effects like this one are invisible due to the absence of structural change in the pocket rather than claiming it applies universally without further examples. revision: partial
-
Referee: [Results (ALK-L1196M analysis)] Results (ALK-L1196M case): The statement that learned embeddings capture affinity-based resistance missed by geometry (Delta_SB = -0.228 versus Delta_P2Rank = -0.95) is presented without controls demonstrating that the effect is not an artifact of the training distribution or specific to the GNN architecture, separate from the reported held-out family generalization.
Authors: The held-out family generalization (AUROC 0.761) provides supporting evidence against training-distribution artifacts, as the model performs well on unseen families. However, we agree that additional controls specific to the ALK-L1196M delta would strengthen the interpretation. We will add a brief discussion in the Results noting this as a potential limitation and clarifying that the embedding effect is consistent with the GNN's learned features beyond geometry alone. revision: partial
-
Referee: [Methods (dataset and evaluation)] Methods (dataset construction and evaluation protocol): The manuscript reports AUROC 0.703 (p=0.026) and held-out AUROC 0.761 on 229 pockets across 25 families, but lacks explicit description of family selection criteria, splice-variant mapping procedure, and safeguards against data leakage that would be required to confirm the generalization claim is load-bearing for the taxonomy.
Authors: We agree that these details are necessary to support the generalization claims. We will revise the Methods section to explicitly describe the criteria used for selecting the 25 kinase families, the procedure for mapping splice variants to pocket structures, and the safeguards against data leakage (including family-level separation between training and test sets). revision: yes
- The small sample of only six clinically validated splice variants (with a single allosteric example) for the resistance taxonomy, which inherently limits the ability to fully distinguish model-specific effects from intrinsic limitations of pocket-centric approaches.
Circularity Check
No significant circularity detected; derivation is self-contained
full rationale
The paper trains a GNN on isoform data and reports AUROC on held-out families (0.761) plus comparisons to P2Rank (p=0.026). The two-tier taxonomy is presented as the outcome of empirical analysis on six variants, with deltas computed from model outputs versus baseline. No equations or steps reduce by construction to fitted inputs, self-definitions, or self-citation chains. The strong claim that allosteric mechanisms are 'fundamentally invisible to any pocket-centric approach' is an interpretive generalization rather than a load-bearing derivation that collapses to the paper's own inputs. The work remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The six variants adequately span the five mechanism classes of splice-mediated resistance.
Reference graph
Works this paper leans on
-
[1]
Ivan A Adzhubei, Steffen Schmidt, Leonid Peshkin, Vasily E Ramensky, Anna Gerasimova, Peer Bork, Alexey S Kondrashov, and Shamil R Sunyaev. 2010. A method and server for predicting damaging missense mutations.Nature Methods 7, 4 (2010), 248–249
2010
-
[2]
Emmanuel S Antonarakis, Changxue Lu, Hao Wang, Brandon Luber, Mary Nakazawa, Jeffrey C Roeser, Yan Chen, Tabrez A Mohammad, Yidong Chen, Helen L Fedor, et al. 2014. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer.New England Journal of Medicine371, 11 (2014), 1028–1038
2014
-
[3]
BioRender. 2024. Created with BioRender.com. https://www.biorender.com. Accessed 2024
2024
-
[4]
Jun Cheng, Guido Novati, Joshua Pan, Clare Bycroft, Akvil˙e Žemgulyt ˙e, Taylor Applebaum, Alexander Pritzel, et al. 2023. Accurate proteome-wide missense variant effect prediction with AlphaMissense.Science381, 6664 (2023), eadg7492
2023
-
[5]
Mindy I Davis, Jeremy P Hunt, Sanna Herrgard, Pietro Ciceri, Lisa M Wodicka, Gabriel Pallares, Michael Hocker, Daniel K Treiber, and Patrick P Zarrinkar. 2011. Comprehensive analysis of kinase inhibitor selectivity.Nature Biotechnology29, 11 (2011), 1046–1051
2011
-
[6]
Heidi Dvinge, Eunhee Kim, Omar Abdel-Wahab, and Robert K Bradley. 2016. RNA splicing factors as oncoproteins and tumour suppressors.Nature Reviews Cancer16, 7 (2016), 413–430
2016
-
[7]
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steineg- ger, Debsindhu Bhowmik, and Burkhard Rost. 2022. ProtTrans: toward under- standing the language of life through self-supervised learning.IEEE Transactions on Pattern Analysis and Machine Intelligence44, 10 ...
2022
-
[8]
Pablo Gainza, Freyr Sverrisson, Federico Monti, Emanuele Rodolà, Davide Boscaini, Michael M Bronstein, and Bruno E Correia. 2020. Deciphering in- teraction fingerprints from protein molecular surfaces using geometric deep learning.Nature Methods17, 2 (2020), 184–192
2020
-
[9]
Anna Gaulton, Louisa J Bellis, A Patricia Bento, Jon Chambers, Mark Davies, Anne Hersey, Yvonne Light, Shaun McGlinchey, David Michalovich, Bissan Al-Lazikani, et al. 2012. ChEMBL: a large-scale bioactivity database for drug discovery.Nucleic Acids Research40, D1 (2012), D1100–D1107
2012
-
[10]
Vladimir Gligorijević, P Douglas Renfrew, Tomasz Kosciolek, Julia Koehler Leman, Daniel Berenberg, Tommi Vatanen, Chris Chandler, Bryn C Taylor, Ian M Fisk, Hera Vlamakis, et al. 2021. Structure-based protein function prediction using graph convolutional networks.Nature Communications12, 1 (2021), 3168
2021
-
[11]
Thomas A Halgren. 2009. Identifying and characterizing binding sites and assessing druggability.Journal of Chemical Information and Modeling49, 2 (2009), 377–389
2009
-
[12]
Aaron N Hata, Matthew J Niederst, Hannah L Archibald, et al. 2016. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition.Nature Medicine22, 3 (2016), 262–269
2016
-
[13]
Andrew L Hopkins and Colin R Groom. 2002. The druggable genome.Nature Reviews Drug Discovery1, 9 (2002), 727–730
2002
-
[14]
José Jiménez, Stefan Doerr, Gerard Martínez-Rosell, Alexander S Rose, and Gianni De Fabritiis. 2017. DeepSite: protein-binding site predictor using 3D- convolutional neural networks.Bioinformatics33, 19 (2017), 3036–3042
2017
-
[15]
José Jiménez, Miha Škalič, Gerard Martínez-Rosell, and Gianni De Fabritiis. 2018. KDEEP: protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks.Journal of Chemical Information and Modeling58, 2 (2018), 287–296
2018
-
[16]
Bowen Jing, Stephan Eismann, Pratham N Soni, and Ron O Dror. 2021. Equi- variant graph neural networks for 3D macromolecular structure. InInternational Conference on Machine Learning (ICML) Workshop on Computational Biology. PMLR, Virtual
2021
-
[17]
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. 2021. Highly accurate protein structure prediction with Al- phaFold.Nature596, 7873 (2021), 583–589
2021
-
[18]
André Kahles, Kjong-Van Lehmann, Nora C Toussaint, Matthias Hüser, Stefan G Stark, Timo Sachsenberg, Oliver Stegle, Oliver Kohlbacher, Chris Sander, Gunnar Rätsch, and Cancer Genome Atlas Research Network. 2018. Comprehensive analysis of alternative splicing across tumors from 8,705 patients.Cancer Cell34, 2 (2018), 211–224
2018
-
[19]
Radoslav Krivak and David Hoksza. 2018. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics10, 1 (2018), 39
2018
-
[20]
Damjan Krstajic, Ljubomir J Buturovic, David E Leahy, and Simon Thomas
-
[21]
Cross-validation pitfalls when selecting and assessing regression and classification models.Journal of Cheminformatics6, 1 (2014), 10
2014
-
[22]
Jack Kyte and Russell F Doolittle. 1982. A simple method for displaying the hydropathic character of a protein.Journal of Molecular Biology157, 1 (1982), 105–132
1982
-
[23]
Vincent Le Guilloux, Peter Schmidtke, and Pierre Tuffery. 2009. fpocket: an open source platform for ligand pocket detection.BMC Bioinformatics10, 1 (2009), 168
2009
-
[24]
Tsung-Yi Lin, Priya Goyal, Ross Girshick, Kaiming He, and Piotr Dollár. 2017. Focal loss for dense object detection. InIEEE International Conference on Computer Vision (ICCV). IEEE, Venice, Italy, 2980–2988
2017
-
[25]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, Allan Dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Salvatore Candido, and Alexander Rives. 2023. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379, 6637 (2023), 1123–1130
2023
-
[26]
Tiqing Liu, Yuhmei Lin, Xin Wen, Robert N Jorissen, and Michael K Gilson. 2007. BindingDB: a web-accessible database of experimentally determined protein– ligand binding affinities.Nucleic Acids Research35, suppl_1 (2007), D198–D201
2007
-
[27]
Savvas K Mylonas, Apostolos Axenopoulos, and Petros Daras. 2021. DeepSurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins.Bioinformatics37, 12 (2021), 1681–1690
2021
-
[28]
Pauline C Ng and Steven Henikoff. 2003. SIFT: predicting amino acid changes that affect protein function.Nucleic Acids Research31, 13 (2003), 3812–3814
2003
-
[29]
Paul K Paik, Alexander Drilon, Pang-Dian Fan, Helena Yu, Natasha Rekhtman, Michelle S Ginsberg, Laetitia Borsu, Nikolaus Schultz, Michael F Berger, Charles M Rudin, and Marc Ladanyi. 2015. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discovery5, 8 (2015), 842–849
2015
-
[30]
Poulikos I Poulikakos, Yogindra Persaud, Manickam Janakiraman, Xiangju Kong, Charles Ng, Gatien Moriceau, Hubing Shi, Mohammad Atefi, Bjoern Titz, May Tal Gabay, et al . 2011. RAF inhibitor resistance is mediated by dimerization of SpliceBind: Isoform-Aware Prediction of Binding Pocket Druggability ACM-BCB ’26, June 30 – July 3, 2026, Calabria, Italy aber...
2011
-
[31]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, and Rob Fergus. 2021. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the National Academy of Sciences 118, 15 (2021), e2016239118
2021
-
[32]
David R Roberts, Volker Bahn, Simone Ciuti, Mark S Boyce, Jane Elith, Gurutzeta Guillera-Arroita, Severin Hauenstein, José J Lahoz-Monfort, Boris Schröder, Wil- fried Thuiller, et al. 2017. Cross-validation strategies for data with temporal, spatial, hierarchical, or phylogenetic structure.Ecography40, 8 (2017), 913–929
2017
-
[33]
Howard I Scher, David Lu, Nicole A Schreiber, Jessica Louw, Ryon P Graf, Hebert A Vargas, Ann Johnson, Adam Jendrisak, et al . 2016. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer.JAMA Oncology2, 11 (2016), 1441–1449
2016
-
[34]
Kristof T Schütt, Pieter-Jan Kindermans, Huziel Enoc Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert Müller. 2017. SchNet: a continuous-filter convolutional neural network for modeling quantum interactions. InAdvances in Neural Information Processing Systems, Vol. 30. Curran Associates, Inc., Long Beach, CA, USA, 992–1002
2017
-
[35]
Joost Schymkowitz, Jesper Borg, François Stricher, Robby Nys, Frederic Rousseau, and Luis Serrano. 2005. The FoldX web server: an online force field.Nucleic Acids Research33, suppl_2 (2005), W382–W388
2005
-
[36]
Hannes Stärk, Octavian-Eugen Ganea, Lagnajit Pattanaik, Regina Barzilay, and Tommi Jaakkola. 2022. EquiBind: geometric deep learning for drug binding structure prediction. InInternational Conference on Machine Learning. PMLR, Baltimore, MD, USA, 20503–20521
2022
-
[37]
Marta M Stepniewska-Dziubinska, Piotr Zielenkiewicz, and Pawel Siedlecki
-
[38]
Scientific Reports10, 1 (2020), 5035
Improving detection of protein-ligand binding sites with 3D segmentation. Scientific Reports10, 1 (2020), 5035
2020
-
[39]
Javier Tapial, Kevin CH Ha, Timothy Sterne-Weiler, André Gohr, Ulrich Braun- schweig, Antonio Hermoso-Pulido, Mathieu Quesnel-Vallières, Jon Permanyer, Reza Sodaei, Yamile Marquez, et al. 2017. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms...
2017
-
[40]
Mihaly Varadi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natassia, Galabina Yordanova, David Yuan, Oana Stroe, Gemma Wood, Agata Laydon, et al. 2022. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research50, D1 (2022), D439–D444
2022
-
[41]
Andrea Volkamer, Daniel Kuhn, Friedrich Rippmann, and Matthias Rarey. 2012. DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment.Bioinformatics28, 15 (2012), 2074–2075
2012
-
[42]
Yue Wang, Yongbin Sun, Ziwei Liu, Sanjay E Sarma, Michael M Bronstein, and Justin M Solomon. 2019. Dynamic graph CNN for learning on point clouds.ACM Transactions on Graphics38, 5 (2019), 1–12
2019
-
[43]
Jürgen Wolf, Takashi Seto, Ji-Youn Han, Noemi Reguart, Edward B Garon, Harry J M Groen, Daniel S W Tan, Toyoaki Hida, Maja de Jonge, Sergey V Orlov, et al
-
[44]
Capmatinib in MET Exon 14–Mutated or MET-Amplified Non–Small-Cell Lung Cancer.New England Journal of Medicine383, 10 (2020), 944–957
2020
-
[45]
Liangzhen Zheng, Jingrong Fan, and Yuguang Mu. 2019. OnionNet: a multiple- layer intermolecular-contact-based convolutional neural network for protein– ligand binding affinity prediction.ACS Omega4, 14 (2019), 15956–15965. A Full Per-Family Cross-Validation Results Table 4 reports AUROC and AUPRC for each kinase family across all three random seeds. Table...
2019
-
[46]
uses the Kyte-Doolittle hydropathy scale [21], min-max normal- ized from the raw range [−4.5, 4.5] to [0, 1].Net charge(index 21) encodes electrostatic character at physiological pH 7.4: negatively charged residues (D, E) are mapped to 0.0, neutral residues to 0.5, and positively charged residues (K, R) to 1.0.Polarity(index 22) is a binary indicator: pol...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.