On the Promises and Limits of Multi-omics Integration for Deconvolution: The HADACA3 Benchmark
Pith reviewed 2026-06-27 22:54 UTC · model grok-4.3
The pith
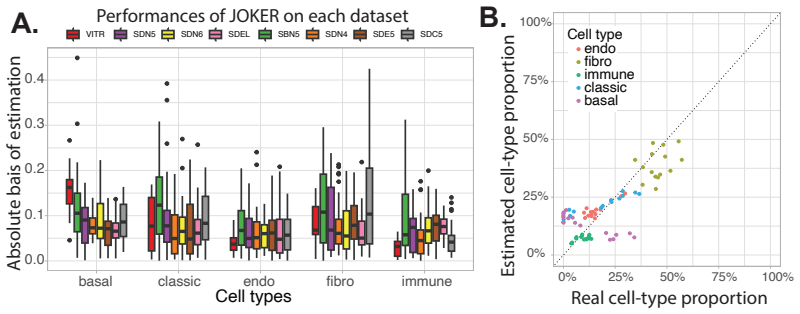
DNA methylation alone achieves the highest median performance across nine datasets for cell-type deconvolution.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
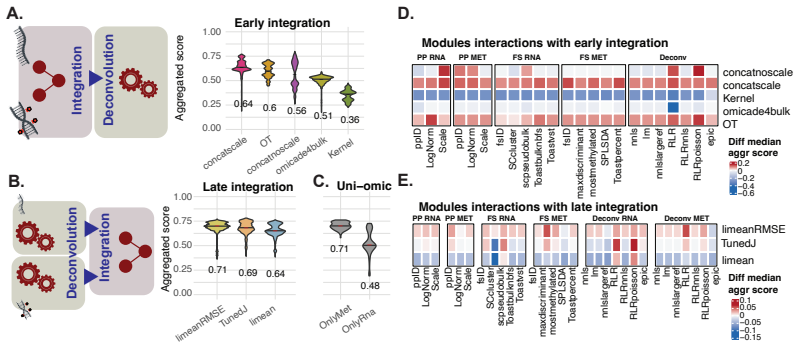
DNAm alone achieves the highest median performance across datasets, making it the most stable and reliable single-modality approach. Multi-omics integration strategies can regularly achieve higher top performance in specific datasets and pipeline configurations. Late integration based on error-weighted averaging provides a strong and reliable baseline, while non-linear early integration methods such as optimal transport show promising results on real biological datasets. Multi-omics integration does not systematically improve average performance over DNAm alone but can improve best-case performance in specific settings.
What carries the argument
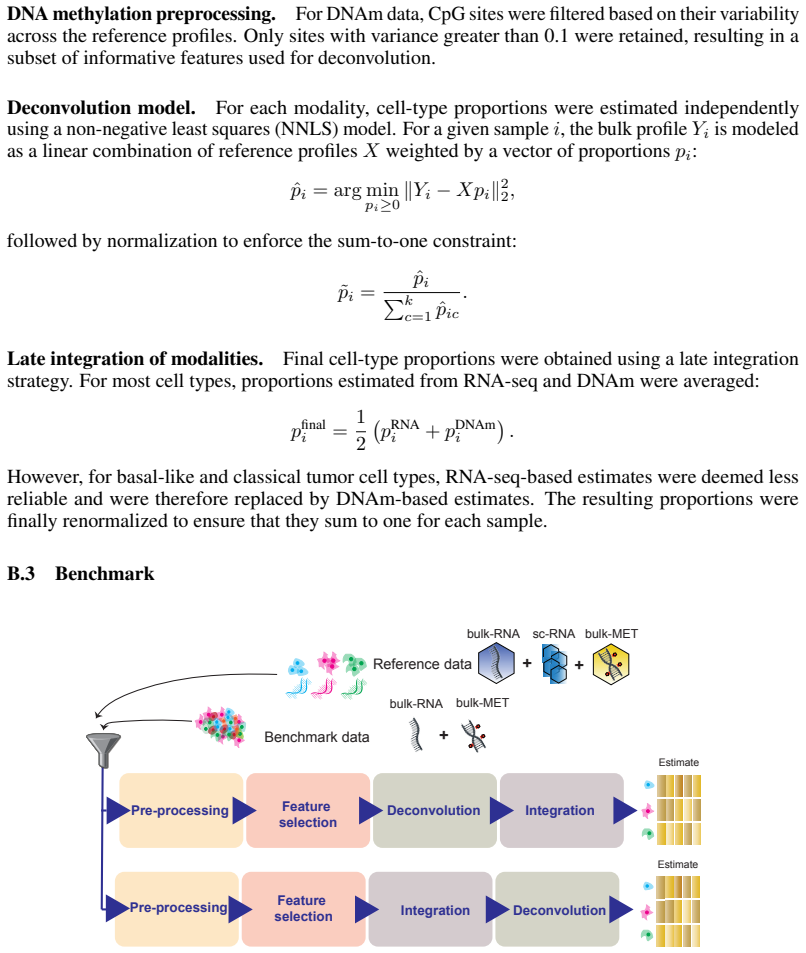
The HADACA3 benchmark that jointly tests preprocessing, feature selection, modeling, and integration strategies on matched DNAm and RNA profiles.
If this is right
- DNAm alone serves as the default reliable choice for most deconvolution tasks.
- Late integration via error-weighted averaging offers consistent gains without sacrificing robustness.
- Non-linear early integration such as optimal transport can deliver superior results on certain real biological datasets.
- Integration strategies must be matched to the statistical properties of each dataset to realize peak performance.
- A trade-off exists between median robustness and occasional best-case gains when adding RNA data.
Where Pith is reading between the lines
- Benchmark designers could add datasets with greater technical variation to test whether the DNAm advantage holds under noisier conditions.
- Method developers might build adaptive pipelines that default to DNAm and selectively incorporate RNA only when it improves a validation metric.
- The observed trade-off suggests prioritizing median stability for clinical applications where consistent performance matters more than occasional peaks.
- Similar benchmarks on additional modalities such as proteomics could reveal whether the same pattern of single-modality stability repeats.
Load-bearing premise
The nine datasets with matched DNAm and RNA profiles are representative of the broader range of biological and experimental conditions in tumor and tissue deconvolution tasks.
What would settle it
Finding a new set of matched DNAm and RNA samples where a majority of multi-omics pipelines exceed DNAm-alone median performance across repeated runs would falsify the stability claim.
Figures









read the original abstract
Understanding the cellular composition of complex tissues, such as tumors, is a key challenge in biology and medicine. A common approach, known as deconvolution, aims to estimate the cellular composition from bulk molecular measurements. With the growing availability of multiple types of molecular data, it is often assumed that combining data sources should improve deconvolution performance. Here, we present HADACA3, a community-driven benchmark designed to evaluate this assumption. We conducted a four-day collaborative competition followed by a large-scale computational benchmark, testing more than 250,000 analysis pipelines across nine datasets with matched DNA methylation (DNAm) and RNA profiles, representing a wide range of biological and experimental conditions. Our framework jointly evaluates the impact of preprocessing, feature selection, modeling, and integration strategies. We find that DNAm alone achieves the highest median performance across datasets, making it the most stable and reliable single-modality approach. However, multi-omics integration strategies can regularly achieve higher top performance in specific datasets and pipeline configurations. Among the tested strategies, late integration based on error-weighted averaging provides a strong and reliable baseline, while non-linear early integration methods, such as optimal transport, show promising results on real biological datasets. Overall, our results show that multi-omics integration does not systematically improve average performance over DNAm alone, but can improve best-case performance in specific settings. This highlights a trade-off between robustness and peak performance, and emphasizes the importance of aligning integration strategies with the statistical properties of the data. All data, code, and evaluation tools are publicly available to support reproducible research and future method development.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the HADACA3 benchmark, a community-driven evaluation of multi-omics integration for cellular deconvolution. It tests over 250,000 analysis pipelines across nine datasets with matched DNA methylation (DNAm) and RNA profiles, assessing the effects of preprocessing, feature selection, modeling, and integration strategies. The central empirical claim is that DNAm alone achieves the highest median performance across datasets and is thus the most stable single-modality approach, while multi-omics integration can yield higher peak performance in specific datasets and configurations; late integration via error-weighted averaging is identified as a strong baseline, and non-linear early integration methods show promise on real data. All data, code, and tools are made public.
Significance. If the results hold, the work supplies a large-scale, reproducible empirical assessment of multi-omics strategies in deconvolution, documenting a robustness-versus-peak-performance trade-off that can inform method selection. The scale of the automated evaluation, the community competition format, and the explicit public release of data, code, and evaluation tools are concrete strengths that support future method development and reproducibility in computational biology.
major comments (1)
- Abstract: The claim that the nine datasets 'represent a wide range of biological and experimental conditions' and thereby establish DNAm as 'the most stable and reliable single-modality approach' is not accompanied by selection criteria, tissue-type coverage statistics, platform diversity, sample-size distribution, or explicit comparison against known sources of variability (e.g., rare-cell mixtures or batch effects). With n=9, the median-performance ranking is sensitive to the particular conditions sampled; without this justification the generalization does not follow from the reported numbers.
Simulated Author's Rebuttal
We thank the referee for the constructive comment on the abstract. We address it below and will revise the manuscript to improve clarity and qualification of claims.
read point-by-point responses
-
Referee: [—] Abstract: The claim that the nine datasets 'represent a wide range of biological and experimental conditions' and thereby establish DNAm as 'the most stable and reliable single-modality approach' is not accompanied by selection criteria, tissue-type coverage statistics, platform diversity, sample-size distribution, or explicit comparison against known sources of variability (e.g., rare-cell mixtures or batch effects). With n=9, the median-performance ranking is sensitive to the particular conditions sampled; without this justification the generalization does not follow from the reported numbers.
Authors: We agree the abstract statement would benefit from supporting details and qualification. The nine datasets were chosen solely on the criterion of public availability of matched DNAm and RNA profiles (from GEO and similar repositories) to enable direct multi-omics comparison; they span blood, adipose, brain, and tumor tissues with Illumina and other platforms and sample sizes ranging from dozens to hundreds. In the revision we will (i) add a supplementary table with explicit selection criteria, tissue/platform/sample-size statistics, and (ii) revise the abstract and discussion to state that DNAm showed the highest median within these available matched datasets rather than claiming broad stability across all conditions. We will also note that rare-cell mixtures and strong batch effects were outside the current scope and that results with n=9 are descriptive of the sampled conditions; future work could expand coverage. These changes qualify the generalization without altering the empirical findings. revision: yes
Circularity Check
Empirical benchmark with direct performance observations; no derivation reduces to inputs
full rationale
The paper reports results from running >250k pipelines on nine fixed datasets and computing observed performance metrics (e.g., median across datasets). The central claim that DNAm alone yields the highest median is a direct summary statistic of those runs, not a fitted parameter renamed as prediction, not a self-defined quantity, and not justified by any self-citation chain. No equations, uniqueness theorems, or ansatzes are invoked that collapse to the paper's own inputs. The representativeness concern raised by the skeptic is an external-validity issue, not a circularity issue.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
A robust workflow to benchmark deconvolution of multi-omic data, November 2024
Elise Amblard, Vadim Bertrand, Luis Martin Pena, Slim Karkar, Florent Chuffart, Mira Ayadi, Aurelia Baures, Lucile Armenoult, Yasmina Kermezli, Jerome Cros, Yuna Blum, and Magali Richard. A robust workflow to benchmark deconvolution of multi-omic data, November 2024. Pages: 2024.11.08.622633 Section: New Results
2024
-
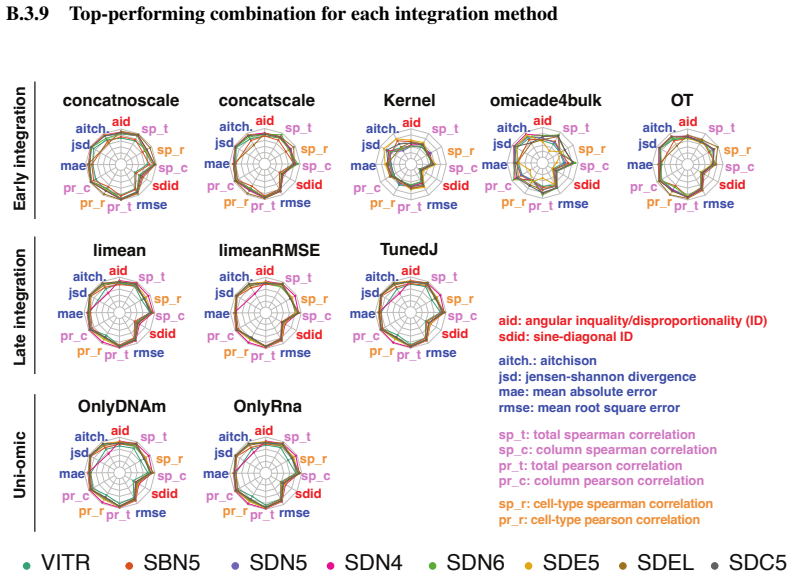
[2]
Powell, Pieter Mestdagh, and Katleen De Preter
Francisco Avila Cobos, José Alquicira-Hernandez, Joseph E. Powell, Pieter Mestdagh, and Katleen De Preter. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nature Communications, 11(1):5650, November 2020. Number: 1
2020
-
[3]
A technical review of multi-omics data integration methods: from classical statistical to deep generative approaches
Ana R Baião, Zhaoxiang Cai, Rebecca C Poulos, Phillip J Robinson, Roger R Reddel, Qing Zhong, Susana Vinga, and Emanuel Gonçalves. A technical review of multi-omics data integration methods: from classical statistical to deep generative approaches. Briefings in Bioinformatics, 26(4):bbaf355, July 2025
2025
-
[4]
Wolock, Aubrey L
Maayan Baron, Adrian Veres, Samuel L. Wolock, Aubrey L. Faust, Renaud Gaujoux, Amedeo Vetere, Jennifer Hyoje Ryu, Bridget K. Wagner, Shai S. Shen-Orr, Allon M. Klein, Douglas A. Melton, and Itai Yanai. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Systems, 3(4):346–360.e4, October 2016
2016
-
[5]
Bastounis, Yi-Ting Yeh, and Julie A
Effie E. Bastounis, Yi-Ting Yeh, and Julie A. Theriot. Subendothelial stiffness alters endothelial cell traction force generation while exerting a minimal effect on the transcriptome. Scientific Reports, 9(1):18209, December 2019. Publisher: Nature Publishing Group
2019
-
[6]
Giraldo, Laetitia Lacroix, Bénédicte Buttard, Nabila Elarouci, Florent Petitprez, Janick Selves, Pierre Laurent-Puig, Catherine Sautès-Fridman, Wolf H
Etienne Becht, Nicolas A. Giraldo, Laetitia Lacroix, Bénédicte Buttard, Nabila Elarouci, Florent Petitprez, Janick Selves, Pierre Laurent-Puig, Catherine Sautès-Fridman, Wolf H. Fridman, and Aurélien de Reyniès. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biology, 17(1):1–20,...
2016
-
[7]
Poulos, Jia Liu, and Qing Zhong
Zhaoxiang Cai, Rebecca C. Poulos, Jia Liu, and Qing Zhong. Machine learning for multi-omics data integration in cancer. iScience, 25(2):103798, February 2022
2022
-
[8]
A unified computational framework for single-cell data integration with optimal transport
Kai Cao, Qiyu Gong, Yiguang Hong, and Lin Wan. A unified computational framework for single-cell data integration with optimal transport. Nature Communications, 13(1):7419, December 2022
2022
-
[9]
Tim H. H. Coorens, Amy Guillaumet-Adkins, Rothem Kovner, Rebecca L. Linn, Victoria H. J. Roberts, Amrita Sule, and Patrick M. Van Hoose. The human and non-human primate developmental GTEx projects. Nature, 637(8046):557–564, January 2025. Publisher: Nature Publishing Group
2025
-
[10]
Benchmarking of methods for DNA methylome deconvolution
Kobe De Ridder, Huiwen Che, Kaat Leroy, and Bernard Thienpont. Benchmarking of methods for DNA methylome deconvolution. Nature Communications, 15(1):4134, May 2024. 10
2024
-
[11]
DECON- bench: a benchmarking platform dedicated to deconvolution methods for tumor heterogeneity quantification
Clémentine Decamps, Alexis Arnaud, Florent Petitprez, Mira Ayadi, Aurélia Baurès, Lucile Armenoult, HADACA consortium, Sergio Escalera, Isabelle Guyon, Rémy Nicolle, Richard Tomasini, Aurélien de Reyniès, Jérôme Cros, Yuna Blum, and Magali Richard. DECON- bench: a benchmarking platform dedicated to deconvolution methods for tumor heterogeneity quantificat...
2021
-
[12]
Computational deconvolution of DNA methylation data from mixed DNA samples
Maísa R Ferro dos Santos, Edoardo Giuili, Andries De Koker, Celine Everaert, and Katleen De Preter. Computational deconvolution of DNA methylation data from mixed DNA samples. Briefings in Bioinformatics, 25(3):bbae234, May 2024
2024
-
[13]
Julia Franzen, Anne Zirkel, Jonathon Blake, Björn Rath, Vladimir Benes, Argyris Papantonis, and Wolfgang Wagner. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell, 16(1):183–191, 2017. _eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1111/acel.12544
-
[14]
A Factor Model Ap- proach to Multiple Testing Under Dependence
Chloé Friguet, Kloareg , Maela, , and David Causeur. A Factor Model Ap- proach to Multiple Testing Under Dependence. Journal of the American Statistical Association, 104(488):1406–1415, December 2009. Publisher: ASA Website _eprint: https://doi.org/10.1198/jasa.2009.tm08332
-
[15]
HADACA consortium, Clémentine Decamps, Florian Privé, Raphael Bacher, Daniel Jost, Arthur Waguet, Eugene Andres Houseman, Eugene Lurie, Pavlo Lutsik, Aleksandar Milosavljevic, Michael Scherer, Michael G. B. Blum, and Magali Richard. Guidelines for cell-type hetero- geneity quantification based on a comparative analysis of reference-free DNA methylation de...
2020
-
[16]
Hadaca3 benchmark: Multi-omic deconvolution challenge
HADACA3 Consortium. Hadaca3 benchmark: Multi-omic deconvolution challenge. https: //www.codabench.org/competitions/4714/, 2024. Online benchmark hosted on Cod- abench
2024
-
[17]
Hadaca3 framework: Modular nextflow pipeline for multi-omic deconvolution benchmarking, 2026
HADACA3 Consortium. Hadaca3 framework: Modular nextflow pipeline for multi-omic deconvolution benchmarking, 2026. https://anonymous.4open.science/r/hadaca3_ framework-08D7 - Will be released upon acceptance
2026
-
[18]
Hadaca3 in silico multi-omic benchmark datasets, 2026
HADACA3 Consortium. Hadaca3 in silico multi-omic benchmark datasets, 2026. Zenodo repository : https://zenodo.org/records/19677979
-
[19]
Halbrook, Costas A
Christopher J. Halbrook, Costas A. Lyssiotis, Marina Pasca Di Magliano, and Anirban Maitra. Pancreatic cancer: Advances and challenges. Cell, 186(8):1729–1754, April 2023
2023
-
[20]
Hallmarks of cancer—Then and now, and beyond
Douglas Hanahan. Hallmarks of cancer—Then and now, and beyond. Cell, 189(8):2254–2277, April 2026
2026
-
[21]
Accomando, Devin C
Eugene Andres Houseman, William P. Accomando, Devin C. Koestler, Brock C. Christensen, Carmen J. Marsit, Heather H. Nelson, John K. Wiencke, and Karl T. Kelsey. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13(1):86, May 2012
2012
-
[22]
Toast: improving reference-free cell composition estimation by cross-cell type differential analysis
Ziyi Li and Hao Wu. Toast: improving reference-free cell composition estimation by cross-cell type differential analysis. Genome Biology, 20(1):18, 2019
2019
-
[23]
Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes
Gwen Lomberk, Yuna Blum, Rémy Nicolle, Asha Nair, Krutika Satish Gaonkar, Laetitia Marisa, Angela Mathison, Zhifu Sun, Huihuang Yan, Nabila Elarouci, et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nature communications, 9(1):1978, 2018
1978
-
[24]
Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems
Kim-Anh Lê Cao, Simon Boitard, and Philippe Besse. Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinformatics, 12(1):253, June 2011
2011
-
[25]
Unsupervised multiple kernel learning for heterogeneous data integration
Jérôme Mariette and Nathalie Villa-Vialaneix. Unsupervised multiple kernel learning for heterogeneous data integration. Bioinformatics, 34(6):1009–1015, March 2018. 11
2018
-
[26]
Culhane, and Amin Moghaddas Gholami
Chen Meng, Bernhard Kuster, Aedín C. Culhane, and Amin Moghaddas Gholami. A multivariate approach to the integration of multi-omics datasets. BMC bioinformatics, 15:162, May 2014
2014
-
[27]
Mullen and Ivo H
Katharine M. Mullen and Ivo H. M. Van Stokkum. nnls: The Lawson-Hanson Algorithm for Non-Negative Least Squares (NNLS), October 2007. Institution: Comprehensive R Archive Network Pages: 1.6
2007
-
[28]
Suh, Dong-gi Lee, Manu Shivakumar, Matthew E
Yonghyun Nam, Jaesik Kim, Sang-Hyuk Jung, Jakob Woerner, Erica H. Suh, Dong-gi Lee, Manu Shivakumar, Matthew E. Lee, and Dokyoon Kim. Harnessing AI in Multi-Modal Omics Data Integration: Paving the Path for the Next Frontier in Precision Medicine. Annual review of biomedical data science, 7(1):225–250, August 2024
2024
-
[29]
Generation of multi-omic datasets using high-throughput molecular profiling of rna data in human pancreatic cancer (pdac)
NCBI Gene Expression Omnibus. Generation of multi-omic datasets using high-throughput molecular profiling of rna data in human pancreatic cancer (pdac). https://www.ncbi.nlm. nih.gov/geo/query/acc.cgi?acc=GSE328792, 2026. NCBI Gene Expression Omnibus (GEO) GSE328792
2026
-
[30]
Robust enumeration of cell subsets from tissue expression profiles
Aaron M Newman, Chih Long Liu, Michael R Green, Andrew J Gentles, Weiguo Feng, Yue Xu, Chuong D Hoang, Maximilian Diehn, and Ash A Alizadeh. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods, 12(5):453–457, May 2015
2015
-
[31]
Establishment of a pancreatic adenocar- cinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer
Rémy Nicolle, Yuna Blum, Pauline Duconseil, Charles Vanbrugghe, Nicolas Brandone, Flora Poizat, Julie Roques, Martin Bigonnet, Odile Gayet, Marion Rubis, Nabila Elarouci, Lucile Armenoult, Mira Ayadi, Aurélien de Reyniès, Marc Giovannini, Philippe Grandval, Stephane Garcia, Cindy Canivet, Jérôme Cros, Barbara Bournet, Vincent Moutardier, Marine Gilabert, ...
2020
-
[32]
Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts.Cell Reports, 21(9):2458–2470, November 2017
Rémy Nicolle, Yuna Blum, Laetitia Marisa, Celine Loncle, Odile Gayet, Vincent Moutardier, Olivier Turrini, Marc Giovannini, Benjamin Bian, Martin Bigonnet, Marion Rubis, Nabila Elarouci, Lucile Armenoult, Mira Ayadi, Pauline Duconseil, Mohamed Gasmi, Mehdi Ouaissi, Aurélie Maignan, Gwen Lomberk, Jean-Marie Boher, Jacques Ewald, Erwan Bories, Jonathan Garn...
2017
-
[33]
Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma
Junya Peng, Bao-Fa Sun, Chuan-Yuan Chen, Jia-Yi Zhou, Yu-Sheng Chen, Hao Chen, Lulu Liu, Dan Huang, Jialin Jiang, Guan-Shen Cui, Ying Yang, Wenze Wang, Dan Guo, Menghua Dai, Junchao Guo, Taiping Zhang, Quan Liao, Yi Liu, Yong-Liang Zhao, Da-Li Han, Yupei Zhao, Yun-Gui Yang, and Wenming Wu. Single-cell RNA-seq highlights intra-tumoral heterogeneity and mal...
2019
-
[34]
Simul- taneous enumeration of cancer and immune cell types from bulk tumor gene expression data
Julien Racle, Kaat de Jonge, Petra Baumgaertner, Daniel E Speiser, and David Gfeller. Simul- taneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife, 6:e26476, November 2017
2017
-
[35]
Winter, Andrew W
Srivatsan Raghavan, Peter S. Winter, Andrew W. Navia, Hannah L. Williams, Alan DenAdel, Kristen E. Lowder, Jennyfer Galvez-Reyes, Radha L. Kalekar, Nolawit Mulugeta, Kevin S. Kapner, Manisha S. Raghavan, Ashir A. Borah, Nuo Liu, Sara A. Väyrynen, Andressa Dias Costa, Raymond W. S. Ng, Junning Wang, Emma K. Hill, Dorisanne Y . Ragon, Lauren K. Brais, Alex ...
2021
-
[36]
Rashid, Xianlu L
Naim U. Rashid, Xianlu L. Peng, Chong Jin, Richard A. Moffitt, Keith E. V olmar, Brian A. Belt, Roheena Z. Panni, Timothy M. Nywening, Silvia G. Herrera, Kristin J. Moore, Sarah G. Hen- nessey, Ashley B. Morrison, Ryan Kawalerski, Apoorve Nayyar, Audrey E. Chang, Benjamin Schmidt, Hong Jin Kim, David C. Linehan, and Jen Jen Yeh. Purity Independent Subtypi...
2020
-
[37]
Pacpaint: a histology-based deep learning model uncovers the extensive intratumor molecular heterogeneity of pancreatic adenocarcinoma
Charlie Saillard, Flore Delecourt, Benoit Schmauch, Olivier Moindrot, Magali Svrcek, Armelle Bardier-Dupas, Jean Francois Emile, Mira Ayadi, Vinciane Rebours, Louis de Mestier, Pascal Hammel, Cindy Neuzillet, Jean Baptiste Bachet, Juan Iovanna, Nelson Dusetti, Yuna Blum, Magali Richard, Yasmina Kermezli, Valerie Paradis, Mikhail Zaslavskiy, Pierre Courtio...
2023
-
[38]
CATD: a reproducible pipeline for selecting cell-type deconvolution methods across tissues
Anna Vathrakokoili Pournara, Zhichao Miao, Ozgur Yilimaz Beker, Nadja Nolte, Alvis Brazma, and Irene Papatheodorou. CATD: a reproducible pipeline for selecting cell-type deconvolution methods across tissues. Bioinformatics Advances, 4(1):vbae048, 2024
2024
-
[39]
Approaching the holistic transcriptome—convolution and deconvolution in transcriptomics
Maik Wolfram-Schauerte, Thomas V ogel, Hanati Tuoken, Maria Fälth Savitski, Eric Simon, and Kay Nieselt. Approaching the holistic transcriptome—convolution and deconvolution in transcriptomics. Briefings in Bioinformatics, 26(4):bbaf388, July 2025
2025
-
[40]
Shijie C Zheng, Charles E Breeze, Stephan Beck, Danyue Dong, Tianyu Zhu, Liangxiao Ma, Wei Ye, Guoqing Zhang, and Andrew E Teschendorff. EpiDISH web server: Epigenetic Dissection of Intra-Sample-Heterogeneity with online GUI. Bioinformatics, 36(6):1950–1951, March 2020. 9 Acknowledgments We thank all supporting organizations: GDR BIMMM, Région Auvergne-Rh...
-
[41]
Tumor subtypes: basal/classical inferred using PurIST [36]
35 PRJCA001063 (CRA001160) Mapping: Endothelial → endothelial; B cell, T cell, Macrophage → immune; Stellate, Fibroblast → fibroblasts; Ductal cell type 2 → cancer cells. Tumor subtypes: basal/classical inferred using PurIST [36]
-
[42]
Note: no fibroblasts available
7 Broad Institute Single Cell Portal (SCP1644) Mapping: Endothelial → endothelial; B_Cells, Macrophage, T_NK, T_Regs, DC, pDC_cell → immune; Tumor → cancer cells. Note: no fibroblasts available
-
[43]
quantile
4 GSE84133 Mapping: endothelial → endothelial; macrophage, mast, t_cell → immune; activated_stellate, quiescent_stellate → fibrob- lasts. Note: no cancer cells available. DNAm protocol. The library and data were generated using standard Illumina Protocol(Infinium Methylation EPIC BeadChip). The raw DNA methylation intensity data files (IDAT) were processe...
-
[44]
Simulation of pseudo single-cell RNA-seq data. For each gene g and cell type c, RNA counts at the single-cell level were simulated using a negative binomial distribution: X(c) gj ∼ NB(µgc, θgc), where X(c) gj denotes the expression count of gene g in cell j from cell type c, µgc is the expected expression level, and θgc is the dispersion parameter. The me...
-
[45]
Let µpc be the mean methylation level observed in the bulk reference dataset for probe p and cell type c
Simulation of pseudo single-cell DNA methylation data For DNA methylation data, probe-level methylation values were simulated using a discrete distribution over three possible states representing unmethylated, partially methylated, and fully methylated probes: M(c) pj ∈ {0, 0.5, 1}, where M(c) pj denotes the methylation value for probe p in cell j of cell...
-
[46]
, aki) represents the vector of proportions for the k cell types in sample i
Simulation of mixture proportions For each pseudo-bulk sample i, cell-type proportions were generated using a Dirichlet distribution: Ai ∼ Dir(αreal), where Ai = (a1i, . . . , aki) represents the vector of proportions for the k cell types in sample i
-
[47]
For each sample i, a total of K = 100 cells were allocated across cell types according to the proportion vector Ai = (a1i,
Generation of pseudo-bulk samples Pseudo-bulk expression profiles were obtained by aggregating simulated single cells accord- ing to the sampled proportions. For each sample i, a total of K = 100 cells were allocated across cell types according to the proportion vector Ai = (a1i, . . . , aki). The number of cells assigned to cell type c in sample i is: ni...
-
[48]
Claims Question: Do the main claims made in the abstract and introduction accurately reflect the paper’s contributions and scope? Answer: [Yes] Justification: The main claims are supported by a large-scale empirical benchmark covering more than 250,000 method combinations across nine datasets. Our conclusions, in particular that multi-omic integration doe...
-
[49]
Limitations
Limitations Question: Does the paper discuss the limitations of the work performed by the authors? Answer: [Yes] Justification: see Section 7 Guidelines: • The answer [N/A] means that the paper has no limitation while the answer [No] means that the paper has limitations, but those are not discussed in the paper. • The authors are encouraged to create a se...
-
[50]
Guidelines: • The answer [N/A] means that the paper does not include theoretical results
Theory assumptions and proofs 35 Question: For each theoretical result, does the paper provide the full set of assumptions and a complete (and correct) proof? Answer: [N/A] Justification: the paper does not include theoretical results. Guidelines: • The answer [N/A] means that the paper does not include theoretical results. • All the theorems, formulas, a...
-
[51]
All datasets, preprocessing steps, and evaluation procedures are described in the paper Section 4 and 5 and supplementary materials
Experimental result reproducibility Question: Does the paper fully disclose all the information needed to reproduce the main ex- perimental results of the paper to the extent that it affects the main claims and/or conclusions of the paper (regardless of whether the code and data are provided or not)? Answer: [Yes] Justification: Yes. All datasets, preproc...
-
[52]
The code used to simulate datasets and generate all figures presented in the paper and to reproduce the benchmark is also publicly available on GitHub [17]
Open access to data and code Question: Does the paper provide open access to the data and code, with sufficient instruc- tions to faithfully reproduce the main experimental results, as described in supplemental material? Answer: [Yes] Justification: The datasets are described in Sections 3.2 and Supplementary Material B.1 and available in Zenodo [18] and ...
-
[53]
A detailed description of each method and its configuration is provided in the Supplementary Material (Section B.3)
Experimental setting/details Question: Does the paper specify all the training and test details (e.g., data splits, hyperpa- rameters, how they were chosen, type of optimizer) necessary to understand the results? Answer: [Yes] Justification: The full modular pipeline used to generate and evaluate all method com- binations is publicly available as a reprod...
-
[54]
Experiment statistical significance Question: Does the paper report error bars suitably and correctly defined or other appropriate information about the statistical significance of the experiments? Answer: [Yes] Justification: Our evaluation is based on an exhaustive benchmark covering all method combinations across multiple datasets, rather than stochast...
-
[55]
All experiments were run on a high-performance computing cluster
Experiments compute resources Question: For each experiment, does the paper provide sufficient information on the com- puter resources (type of compute workers, memory, time of execution) needed to reproduce the experiments? Answer: [Yes] Justification: Compute resources are described in Section B.4. All experiments were run on a high-performance computin...
-
[56]
• If the authors answer [No], they should explain the special circumstances that require a deviation from the Code of Ethics
Code of ethics Question: Does the research conducted in the paper conform, in every respect, with the NeurIPS Code of Ethics https://neurips.cc/public/EthicsGuidelines? Answer: [Yes] Justification: Guidelines: • The answer [N/A] means that the authors have not reviewed the NeurIPS Code of Ethics. • If the authors answer [No], they should explain the speci...
-
[57]
Guidelines: • The answer [N/A] means that there is no societal impact of the work performed
Broader impacts Question: Does the paper discuss both potential positive societal impacts and negative societal impacts of the work performed? Answer: [Yes] Justification: see Sections 7 and 8. Guidelines: • The answer [N/A] means that there is no societal impact of the work performed. • If the authors answer [N/A] or [No], they should explain why their w...
-
[58]
The benchmark relies on publicly available biological datasets and simulated data
Safeguards Question: Does the paper describe safeguards that have been put in place for responsible release of data or models that have a high risk for misuse (e.g., pre-trained language models, image generators, or scraped datasets)? Answer: [No], Justification: This work does not involve models or datasets with a high risk of misuse, such as generative ...
-
[59]
Guidelines: • The answer [N/A] means that the paper does not use existing assets
Licenses for existing assets Question: Are the creators or original owners of assets (e.g., code, data, models), used in the paper, properly credited and are the license and terms of use explicitly mentioned and properly respected? 39 Answer: [Yes] Justification: All external datasets and resources used in this work are publicly available and have been ap...
-
[60]
Guidelines: • The answer [N/A] means that the paper does not release new assets
New assets Question: Are new assets introduced in the paper well documented and is the documentation provided alongside the assets? Answer: [Yes] Justification: The datasets and code are made available on GEO NCBI, Codabench, GitHub and Zenodo (links provided upon acceptance). Guidelines: • The answer [N/A] means that the paper does not release new assets...
-
[61]
competition
Crowdsourcing and research with human subjects Question: For crowdsourcing experiments and research with human subjects, does the paper include the full text of instructions given to participants and screenshots, if applicable, as well as details about compensation (if any)? Answer: [N/A] Justification:This paper does not involve human subjects or crowdso...
-
[62]
• Depending on the country in which research is conducted, IRB approval (or equivalent) may be required for any human subjects research
Institutional review board (IRB) approvals or equivalent for research with human subjects Question: Does the paper describe potential risks incurred by study participants, whether such risks were disclosed to the subjects, and whether Institutional Review Board (IRB) approvals (or an equivalent approval/review based on the requirements of your country or ...
-
[63]
Declaration of LLM usage Question: Does the paper describe the usage of LLMs if it is an important, original, or non-standard component of the core methods in this research? Note that if the LLM is used only for writing, editing, or formatting purposes and does not impact the core methodology, scientific rigor, or originality of the research, declaration ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.