Matrix representations and distance metrics for unlabeled ranked phylogenetic networks
Pith reviewed 2026-06-27 18:17 UTC · model grok-4.3
The pith
A bijective triangular matrix representation turns comparisons of ranked phylogenetic networks into standard matrix norm calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Rooted ranked unlabeled phylogenetic networks admit a bijective triangular matrix representation whose entries encode the order of internal events, speciations, and hybridizations; matrix norms on these matrices therefore supply distances that compare topologies, timed networks, and networks with unequal numbers of hybridizations, and that apply equally to isochronous and heterochronous cases.
What carries the argument
bijective triangular matrix representation that captures the temporal order of internal events, speciations, and hybridizations
If this is right
- Network topologies can be compared quantitatively using efficient matrix operations.
- Networks with different numbers of hybridizations become directly comparable.
- Both isochronous and heterochronous networks are handled by the same distance.
- The metrics can be applied to posterior distributions from Bayesian inference of viral networks.
Where Pith is reading between the lines
- The matrix encoding may support clustering or summarization of large sets of inferred networks from Bayesian analyses.
- Branch-length or timing information could be incorporated into the matrix entries in future extensions.
- Analogous triangular representations might be developed for other classes of reticulate graphs.
Load-bearing premise
Every rooted ranked unlabeled phylogenetic network possesses a unique bijective triangular matrix representation that fully encodes the temporal order of its events.
What would settle it
Two distinct rooted ranked unlabeled phylogenetic networks that produce identical triangular matrices, or a valid network that cannot be represented by any such matrix.
Figures
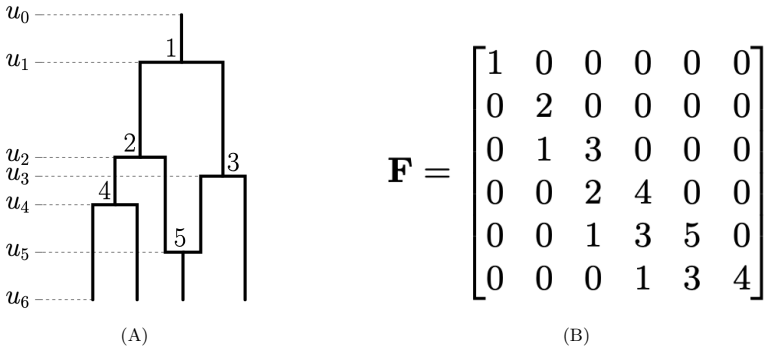
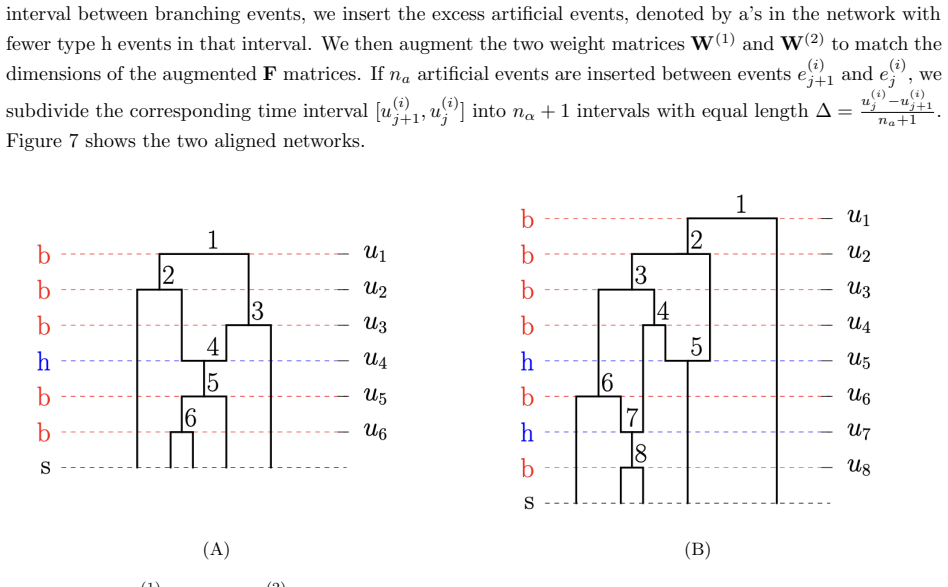

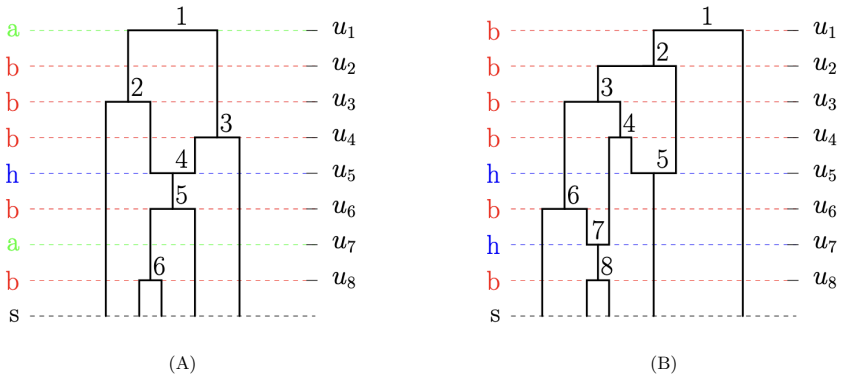
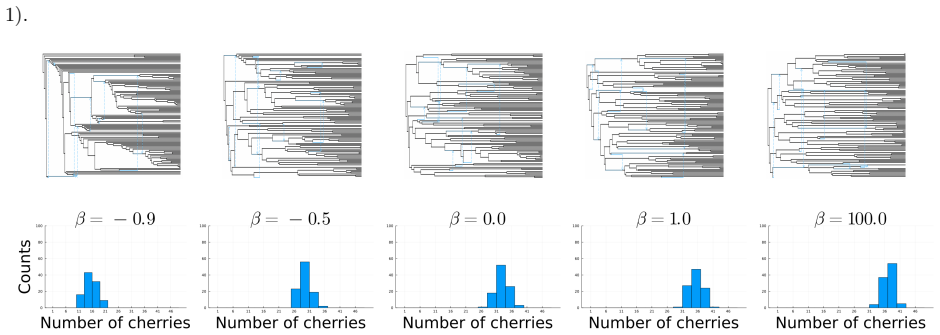
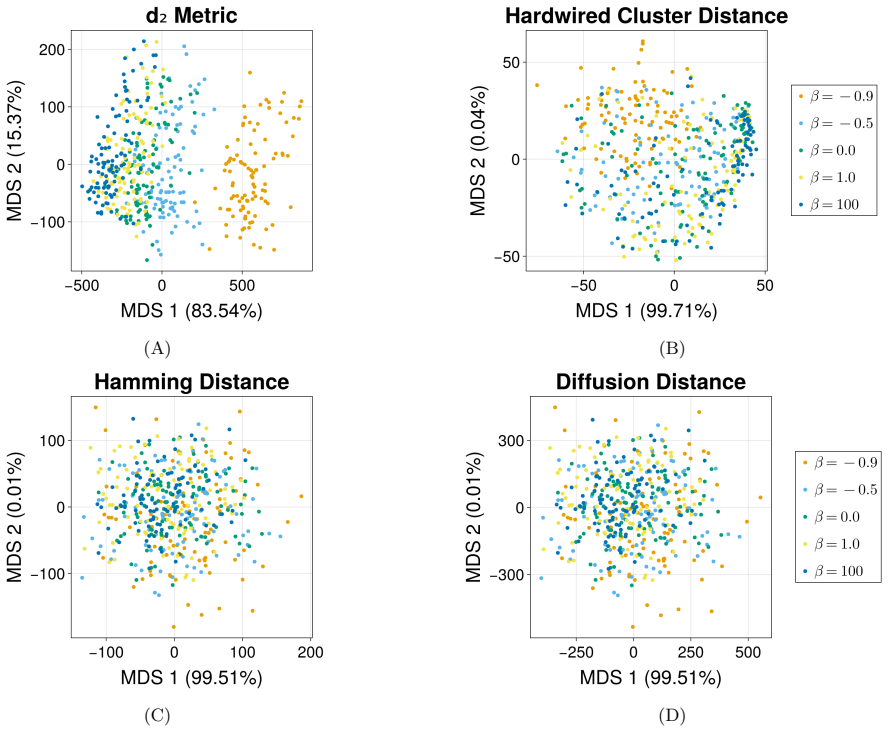
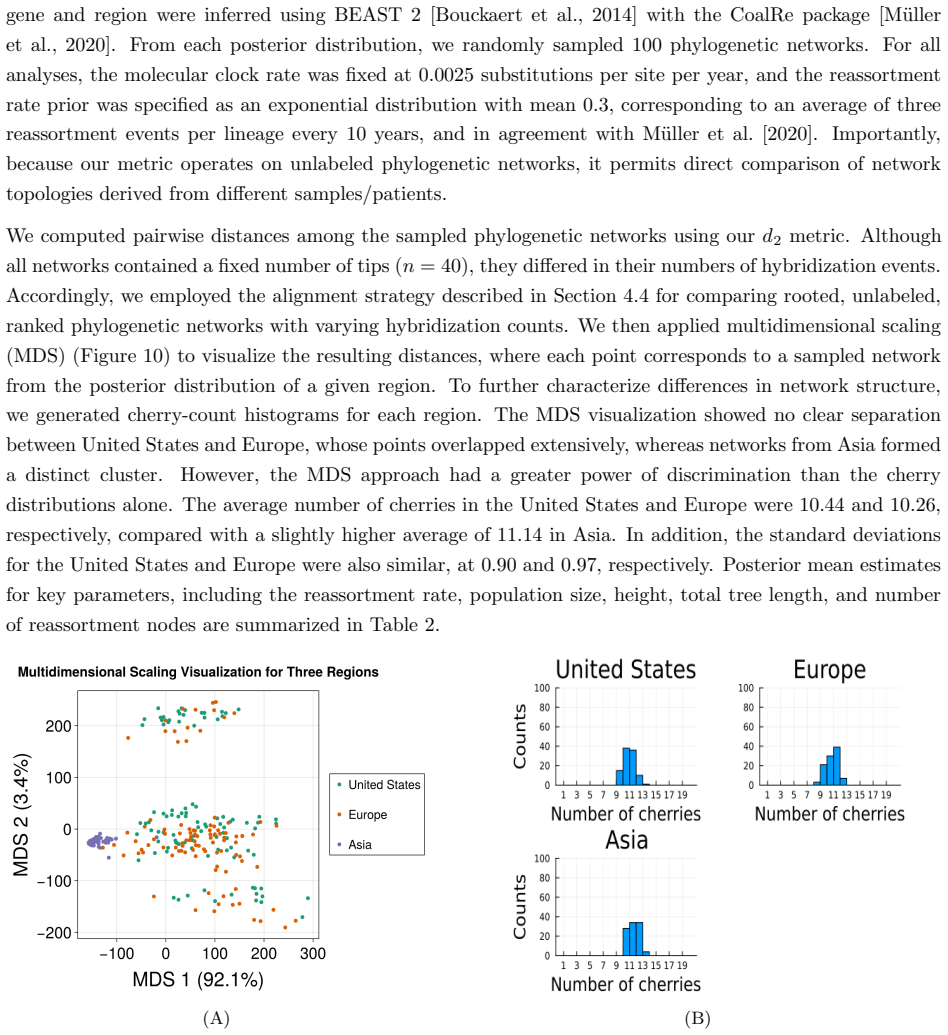
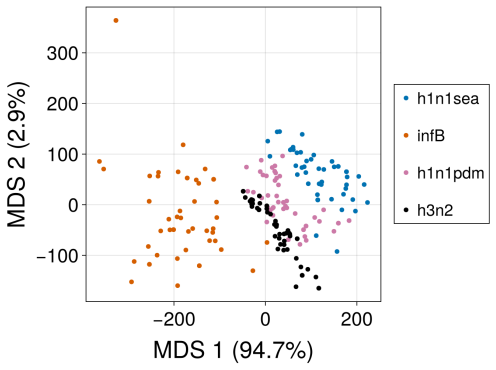
read the original abstract
Phylogenetic networks are graphs inferred from molecular sequence data that represent ancestral histories shaped by reticulate processes such as recombination, hybridization, and horizontal gene transfer. We introduce a family of distance metrics for rooted, ranked, unlabeled phylogenetic networks, extending a previously developed distance for ranked trees. Our approach relies on a bijective triangular matrix representation of phylogenetic networks that captures the temporal order of internal events, speciations, and hybridizations. Our metrics, defined as standard matrix norms, allow efficient quantitative comparisons of network topologies, timed networks and networks with differing numbers of hybridizations. Our distance can be used for both isochronous networks where all tips are sampled at one time point, and heterochronous networks where tips are allowed to be sampled at different time points. We show that our metrics capture biologically meaningful differences among evolutionary histories in both simulations and empirical posterior distributions of viral phylogenetic networks. These tools fill a methodological gap, enabling principled comparisons of ranked, unlabeled phylogenetic networks, including ancestral recombination graphs.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a family of distance metrics for rooted, ranked, unlabeled phylogenetic networks by defining a bijective triangular matrix representation that encodes the temporal order of speciations and hybridizations. Distances are then taken as standard matrix norms, claimed to enable comparisons of topologies, timed networks, and networks with differing reticulation counts for both isochronous and heterochronous cases. Utility is illustrated via simulations and empirical posterior distributions of viral networks.
Significance. If the bijectivity and uniqueness claims hold, the work supplies a concrete, computable framework for quantitative network comparison that extends existing tree metrics and addresses a clear methodological gap; the simulation and empirical demonstrations provide initial evidence of biological relevance.
major comments (2)
- [Abstract] Abstract and introduction: the central claim that a bijective triangular matrix representation exists and uniquely captures temporal order for all rooted ranked unlabeled networks (including variable hybridization counts) is asserted without an explicit construction, injectivity proof, or surjectivity argument; this is load-bearing for the subsequent matrix-norm distances.
- [Abstract] The assertion that the metrics distinguish networks with differing numbers of hybridizations relies on the matrix representation being well-defined and bijective across heterochronous cases, yet no verification or counter-example analysis is supplied to confirm this.
minor comments (1)
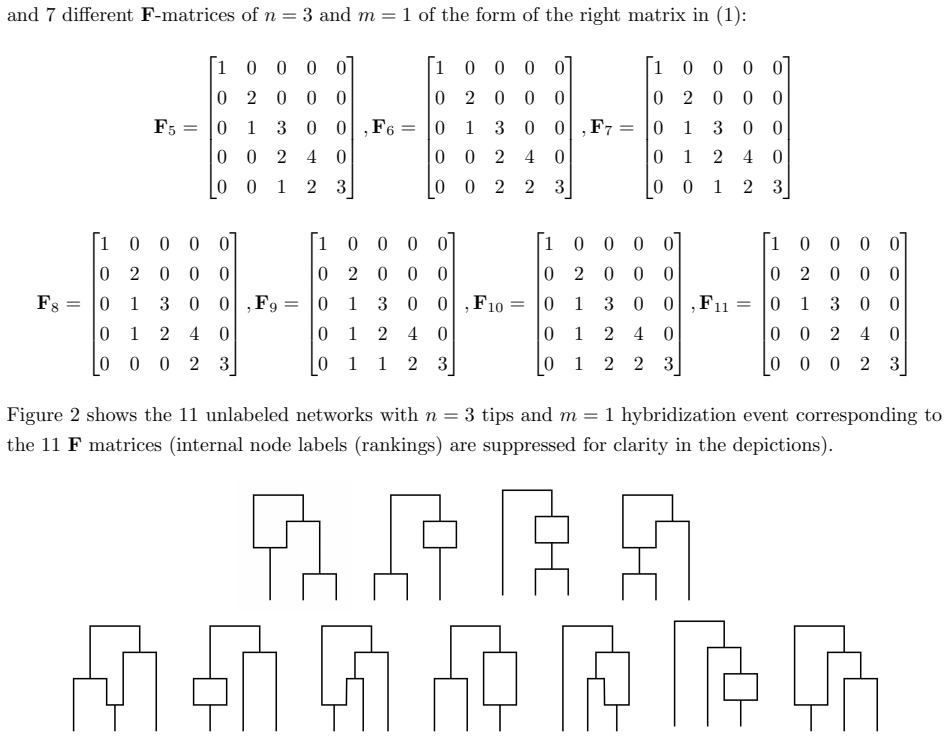
- Notation for the triangular matrix entries and the precise mapping from network events to matrix positions should be defined earlier and with an example.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which help clarify the presentation of our central claims. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract and introduction: the central claim that a bijective triangular matrix representation exists and uniquely captures temporal order for all rooted ranked unlabeled networks (including variable hybridization counts) is asserted without an explicit construction, injectivity proof, or surjectivity argument; this is load-bearing for the subsequent matrix-norm distances.
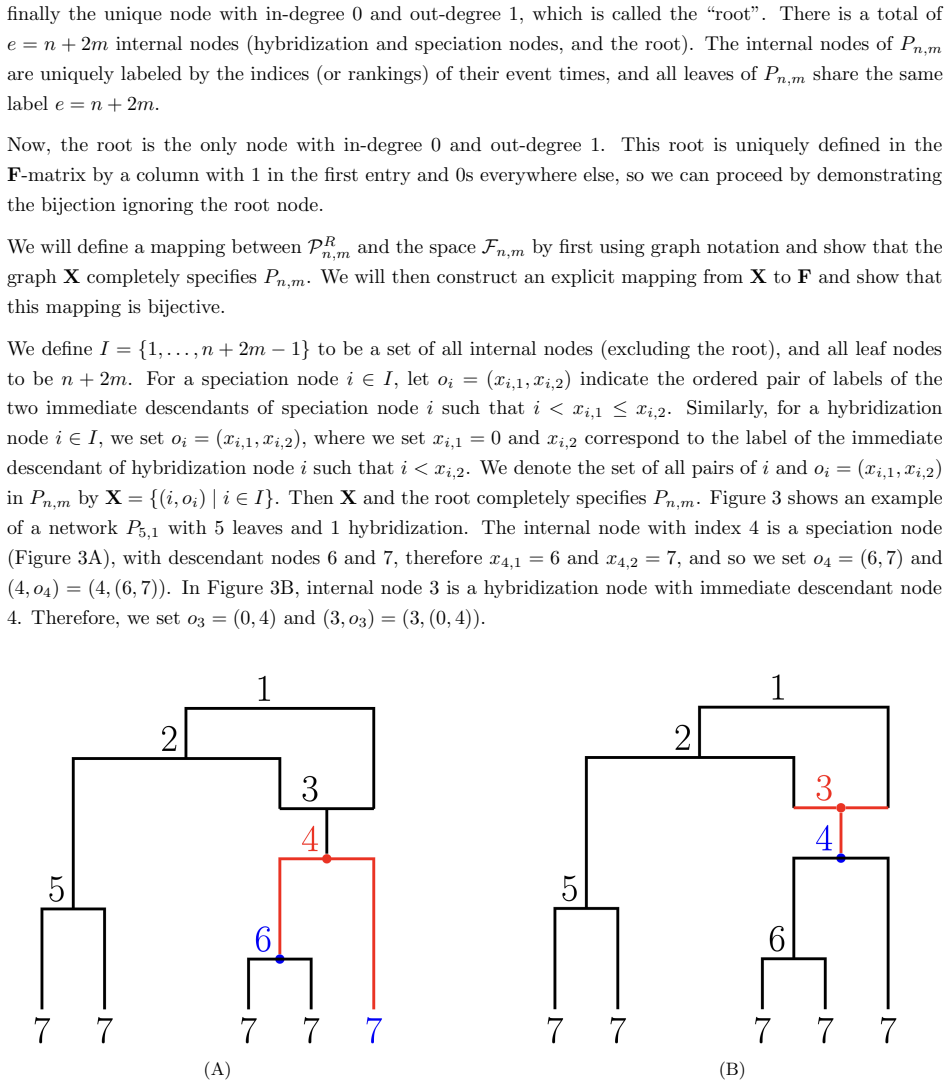
Authors: The explicit construction of the bijective triangular matrix representation appears in Section 2, with the mapping defined via the temporal ordering of internal nodes. Injectivity is established in Theorem 3.1 and surjectivity in Theorem 3.2; both theorems explicitly cover networks with arbitrary reticulation counts. We agree that the abstract and introduction would be strengthened by forward references to these results. We will revise both sections to briefly describe the construction and cite the theorems. revision: yes
-
Referee: [Abstract] The assertion that the metrics distinguish networks with differing numbers of hybridizations relies on the matrix representation being well-defined and bijective across heterochronous cases, yet no verification or counter-example analysis is supplied to confirm this.
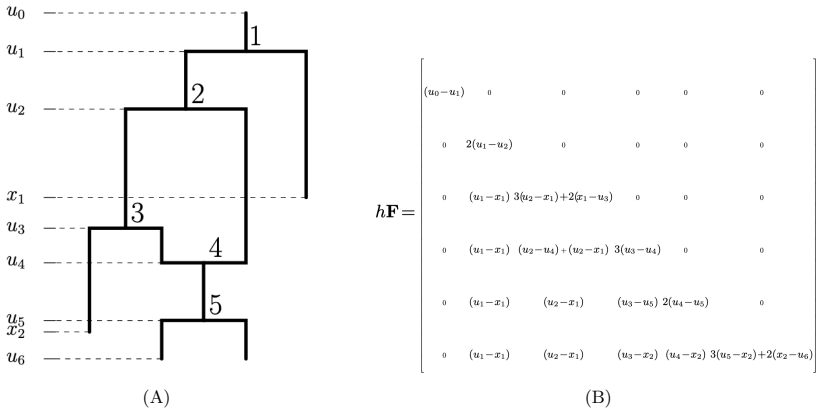
Authors: Section 2.3 defines the representation for heterochronous networks by augmenting the matrix with tip-time information, preserving bijectivity and thereby ensuring distinct reticulation counts map to distinct matrices. While the simulation studies include heterochronous examples, we acknowledge the absence of a dedicated verification subsection. We will add such a subsection containing explicit checks and a short counter-example search confirming that the bijectivity property holds without collision in the heterochronous setting. revision: yes
Circularity Check
No circularity: constructive definitions of matrix representation and matrix-norm distances
full rationale
The paper defines a triangular matrix representation for rooted ranked unlabeled phylogenetic networks and applies standard matrix norms to obtain distances. This is a direct constructive mapping and metric definition, not a derivation that reduces to its own inputs by construction, fitted parameters renamed as predictions, or load-bearing self-citations. The bijectivity claim is presented as a property of the representation rather than an assumption that circularly justifies the distances. No equations or steps in the abstract or description exhibit the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URLhttps://www.pnas.org/doi/abs/10.1073/pnas.1116871109
doi: 10.1073/pnas.1116871109. URLhttps://www.pnas.org/doi/abs/10.1073/pnas.1116871109. David Aldous. Probability distributions on cladograms. In David Aldous and Robin Pemantle, editors, Random Discrete Structures, pages 1–18, New York, NY,
-
[2]
doi: 10.1007/s11538-018-0485-4. Michael G. B. Blum and Olivier Fran¸ cois. Which Random Processes Describe the Tree of Life? A Large-Scale Study of Phylogenetic Tree Imbalance. Systematic Biology, 55(4):685–691, 08
-
[3]
doi: 10.1080/10635150600889625
ISSN 1063-5157. doi: 10.1080/10635150600889625. URLhttps://doi.org/10.1080/10635150600889625. Remco Bouckaert, Joseph Heled, Denise K¨ uhnert, Timothy Vaughan, Chieh-Hsi Wu, Dong Xie, Marc Suchard, Andrew Rambaut, and Alexei Drummond. Beast 2: A software platform for bayesian evolu- tionary analysis. PLoS computational biology, 10:e1003537, 04
-
[4]
Gabriel Cardona, Merc` e Llabr´ es, Francesc Rossell´ o, and Gabriel Valiente
doi: 10.1371/journal.pcbi.1003537. Gabriel Cardona, Merc` e Llabr´ es, Francesc Rossell´ o, and Gabriel Valiente. A distance metric for a class of tree-sibling phylogenetic networks. Bioinformatics, 24(13):1481–1488, 2008a. Gabriel Cardona, Merc` e Llabr´ es, Francesc Rossell´ o, and Gabriel Valiente. Metrics for phylogenetic networks i: Generalizations o...
-
[5]
Neural networks and physical systems with emergent collective com- putational abilities
doi: 10.1073/pnas. 2004999117. Robert C Griffiths and Paul Marjoram. An ancestral recombination graph. Institute for Mathematics and its Applications, 87:257,
-
[6]
Graph diffusion distance: A difference measure for weighted graphs based on the graph laplacian exponential kernel
David Hammond, Yaniv Gur, and Chris Johnson. Graph diffusion distance: A difference measure for weighted graphs based on the graph laplacian exponential kernel. 2013 IEEE Global Conference on Signal and Information Processing, GlobalSIP 2013 - Proceedings, 12
2013
-
[7]
doi: 10.1109/GlobalSIP.2013.6736904. Daniel H. Huson, Regula Rupp, and Celine Scornavacca. Phylogenetic Networks. Cambridge University Press, Cambridge,
-
[8]
ISBN 9780511974076. doi: 10.1017/CBO9780511974076. URLhttp://ebooks. cambridge.org/ref/id/CBO9780511974076. Remie Janssen and Pengyu Liu. Comparing the topology of phylogenetic network generators. Journal of bioinformatics and computational biology, 19(06):2140012,
-
[9]
ISSN 0737-4038. doi: 10.1093/molbev/mst010. URLhttps://doi.org/10.1093/molbev/mst010. Jaehee Kim, Noah Rosenberg, and Julia Palacios. Distance metrics for ranked evolutionary trees.Proceedings of the National Academy of Sciences, 117:28876–28886, 11
-
[10]
Sungsik Kong, Claudia Sol´ ıs-Lemus, and George P Tiley
doi: 10.1073/pnas.1922851117. Sungsik Kong, Claudia Sol´ ıs-Lemus, and George P Tiley. Phylogenetic networks empower biodiversity research. Proceedings of the National Academy of Sciences, 122(31):e2410934122,
-
[11]
doi: 10.1126/science.1250092. Carla Mavian, Sergei Pond, Simone Marini, Brittany Rife Magalis, Anne-Mieke Vandamme, Simon Dellicour, Samuel Scarpino, Charlotte Houldcroft, Christian Julian Villabona-Arenas, Taylor Paisie, N´ ıdia Trov˜ ao, Christina Boucher, Yun Zhang, Richard Scheuermann, Olivier Gascuel, Tommy Lam, Marc Suchard, Ana Abecasis, Eduan Wilk...
-
[12]
Michael Maxfield, Jingcheng Xu, and C´ ecile An´ e
doi: 10.1073/pnas.2007295117. Michael Maxfield, Jingcheng Xu, and C´ ecile An´ e. A dissimilarity measure for semidirected networks.IEEE Transactions on Computational Biology and Bioinformatics,
-
[13]
Vincent Moulton, James Oldman, and Taoyang Wu
doi: 10.1600/036364418X696897. Vincent Moulton, James Oldman, and Taoyang Wu. A cubic-time algorithm for computing the trinet distance between level-1 networks. Information Processing Letters, 123:36–41,
-
[14]
doi: 10.1073/pnas.1918304117. Luay Nakhleh. A metric on the space of reduced phylogenetic networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 7(2):218–222,
-
[15]
24 Origins and evolutionary genomics of the 2009 swine-origin h1n1 influenza a epidemic.Nature, 459:1122–5, 07
Gavin Smith, Vijaykrishna Dhanasekaran, Justin Bahl, Samantha Lycett, Michael Worobey, Oliver Pybus, Siu Ma, Chung Cheung, Jayna Raghwani, Samir Bhatt, Joseph S Peiris, Yi Guan, and Andrew Rambaut. 24 Origins and evolutionary genomics of the 2009 swine-origin h1n1 influenza a epidemic.Nature, 459:1122–5, 07
2009
-
[16]
Claudia Sol´ ıs-Lemus and C´ ecile An´ e
doi: 10.1038/nature08182. Claudia Sol´ ıs-Lemus and C´ ecile An´ e. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genetics, 12(3):e1005896,
-
[17]
Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting
ISSN 1553-7404. doi: 10.1371/journal. pgen.1005896. URLhttp://arxiv.org/abs/1509.06075. Santiago S´ anchez-Pacheco, Sungsik Kong, Paola Pulido-Santacruz, Robert Murphy, and Laura Ku- batko. Median-joining network analysis of sars-cov-2 genomes is neither phylogenetic nor evolutionary. Proceedings of the National Academy of Sciences, 117, 05
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1371/journal
-
[18]
doi: 10.1073/pnas.2007062117. John Wakeley. Coalescent Theory: An Introduction. Roberts & Company Publishers, June
-
[19]
Yun Yu, Jianrong Dong, Kevin J
doi: 10.1371/journal.pgen.1002660. Yun Yu, Jianrong Dong, Kevin J. Liu, and Luay K Nakhleh. Maximum likelihood inference of reticulate evolutionary histories. Proceedings of the National Academy of Sciences of the United States of America, 111 46:16448–53,
-
[20]
URLhttps:// royalsocietypublishing.org/doi/abs/10.1098/rstb.1925.0002
doi: 10.1098/rstb.1925.0002. URLhttps:// royalsocietypublishing.org/doi/abs/10.1098/rstb.1925.0002. Chi Zhang, Huw A. Ogilvie, Alexei J. Drummond, and Tanja Stadler. Bayesian inference of species networks from multilocus sequence data. Molecular Biology and Evolution,
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.