ProHiFlo: Hierarchical Flow Matching with Functional Guidance for De Novo Protein Generation
Pith reviewed 2026-06-28 07:09 UTC · model grok-4.3
The pith
ProHiFlo generates de novo proteins via hierarchical flow matching and functional guidance from pretrained predictors, achieving higher success rates with four fewer sampling steps.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
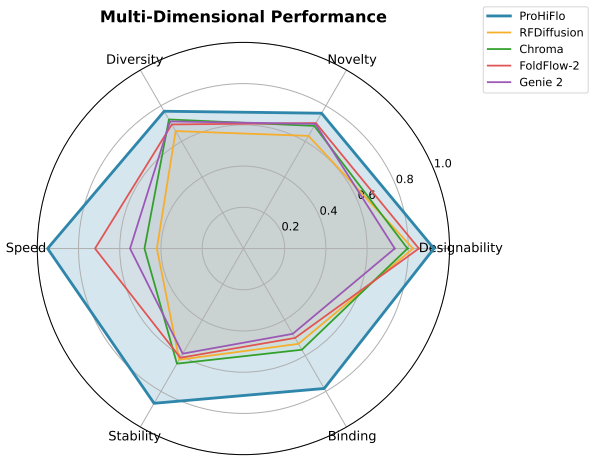
ProHiFlo shows that a hierarchical flow matching process combined with functional guidance from pretrained predictors produces more accurate and efficient de novo proteins than single-resolution diffusion or flow models, delivering state-of-the-art performance on multiple design tasks with substantially reduced sampling steps.
What carries the argument
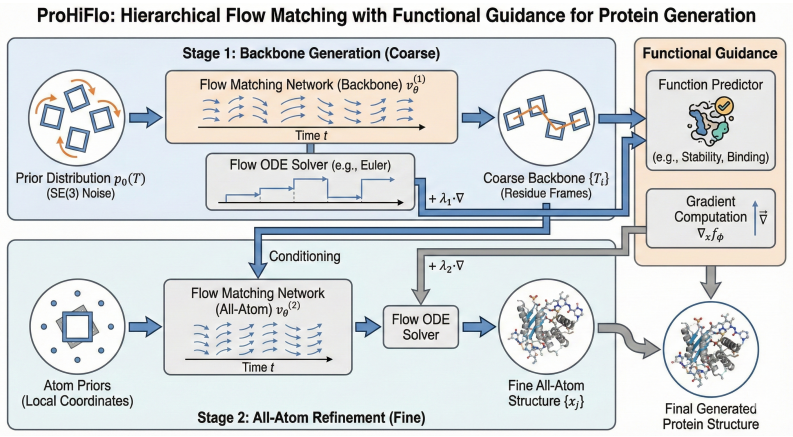
The hierarchical flow matching framework with functional guidance, which generates structures coarse-to-fine and steers them using external pretrained predictors.
If this is right
- Protein design pipelines can run with four fewer sampling steps while maintaining or improving output quality.
- Functional constraints can be added to existing flow matching models without full retraining.
- Multi-scale modeling separates backbone geometry from all-atom refinement to lower computational cost.
- Enzyme active site scaffolding success rises from 41.2 percent to 58.9 percent under the new approach.
Where Pith is reading between the lines
- The same coarse-to-fine structure could be tested on other sequence-to-structure tasks such as antibody design.
- Predictor-based guidance might reduce the need for task-specific fine-tuning across generative biology models.
- Integration with downstream experimental assays could create closed-loop design systems that update guidance signals iteratively.
Load-bearing premise
Functional guidance from pretrained predictors can steer generation toward desired properties without retraining the core model.
What would settle it
A controlled test in which functional guidance is removed and success rates on enzyme active site scaffolding fall to or below the 41.2 percent baseline of prior methods.
Figures


read the original abstract
De novo protein generation has transformative potential in therapeutic design, enzyme engineering, and synthetic biology. While diffusion-based and flow matching approaches have achieved progress, they typically operate at single resolution and lack mechanisms for incorporating functional constraints. We introduce ProHiFlo, a hierarchical flow matching framework with three innovations: (1) coarse-to-fine generation that models backbone geometry before refining to all-atom coordinates, reducing computational cost while maintaining accuracy; (2) functional guidance leveraging pretrained predictors to steer generation toward desired properties without retraining; (3) adaptive SE(3)-equivariant architecture for efficient multi-scale processing. Experiments on unconditional generation, motif scaffolding, and functional design demonstrate state-ofthe-art performance while requiring 4 fewer sampling steps. On enzyme active site scaffolding, ProHiFlo achieves 58.9% success rate compared to 41.2% for RFDiffusion.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces ProHiFlo, a hierarchical flow matching framework for de novo protein generation featuring three innovations: (1) coarse-to-fine modeling of backbone geometry before all-atom refinement, (2) functional guidance from pretrained predictors to steer toward desired properties without retraining the core model, and (3) an adaptive SE(3)-equivariant architecture. It reports state-of-the-art results on unconditional generation, motif scaffolding, and functional design tasks, including a reduction of 4 sampling steps and a 58.9% success rate on enzyme active site scaffolding versus 41.2% for RFDiffusion.
Significance. If the reported performance gains hold under rigorous validation, the work would be significant for computational protein design. The combination of hierarchical flow matching with predictor-based guidance offers a practical route to property-conditioned generation that avoids full retraining, while the reduced sampling steps address a key efficiency bottleneck in diffusion/flow-based methods for biomolecules. The approach extends existing equivariant generative models in a manner that could generalize to other functional constraints.
major comments (2)
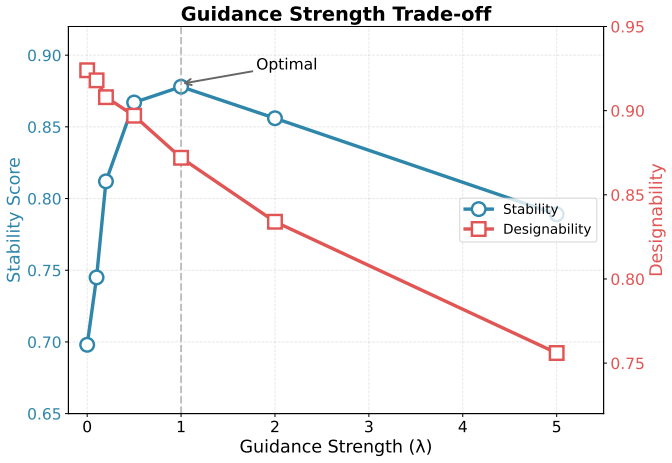
- The central performance claims (e.g., 58.9% vs. 41.2% success on enzyme scaffolding) are attributed in part to functional guidance, yet the manuscript provides no quantitative assessment of how well the pretrained predictors transfer to generated (as opposed to natural) structures; without held-out accuracy, correlation with downstream success metrics, or ablation on guidance strength, it is unclear whether the observed delta can be credited to this component rather than the hierarchical architecture alone.
- The experimental section does not report error bars, number of independent runs, or statistical significance tests for the success-rate comparisons; given that the abstract already highlights specific numerical improvements, the absence of these details makes it impossible to assess whether the gains are robust or within expected variance of the baselines.
minor comments (2)
- Abstract: 'state-ofthe-art' contains a typographical error and should read 'state-of-the-art'.
- The description of the adaptive SE(3)-equivariant architecture would benefit from an explicit statement of how the multi-scale processing differs from prior equivariant flow-matching models (e.g., a brief comparison paragraph).
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which highlight important aspects of validation and statistical rigor. We address each major point below and outline planned revisions.
read point-by-point responses
-
Referee: The central performance claims (e.g., 58.9% vs. 41.2% success on enzyme scaffolding) are attributed in part to functional guidance, yet the manuscript provides no quantitative assessment of how well the pretrained predictors transfer to generated (as opposed to natural) structures; without held-out accuracy, correlation with downstream success metrics, or ablation on guidance strength, it is unclear whether the observed delta can be credited to this component rather than the hierarchical architecture alone.
Authors: We agree that isolating the contribution of functional guidance requires additional analysis. The reported gains reflect the full framework, but the manuscript does not include an explicit ablation on guidance strength or direct transfer metrics for generated structures. In revision we will add an ablation varying guidance strength and report any available correlations between predictor outputs and success rates. A full held-out accuracy evaluation on generated structures was not performed in the original experiments. revision: partial
-
Referee: The experimental section does not report error bars, number of independent runs, or statistical significance tests for the success-rate comparisons; given that the abstract already highlights specific numerical improvements, the absence of these details makes it impossible to assess whether the gains are robust or within expected variance of the baselines.
Authors: We acknowledge the value of reporting variability. The presented success rates follow the single-run protocols used by prior methods such as RFDiffusion, but we will conduct additional independent runs, include error bars, and add statistical significance tests for the key comparisons in the revised manuscript. revision: yes
Circularity Check
No circularity in derivation or claims
full rationale
The paper introduces an empirical ML architecture (hierarchical flow matching plus functional guidance via external pretrained predictors) and supports its performance claims through direct experimental comparisons against independent baselines such as RFDiffusion. No equations, predictions, or uniqueness arguments are shown to reduce to self-definitions, fitted inputs renamed as outputs, or load-bearing self-citations. The functional-guidance step relies on an external assumption about predictor transfer rather than any internal circular reduction. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
The coming of age of de novo protein design
Po-Ssu Huang, Scott E Boyken, and David Baker. The coming of age of de novo protein design. Nature, 537(7620):320–327, 2016
2016
-
[2]
Advanced deep learning methods for protein structure prediction and design.BIO Integration, 2025
Yichao Zhang, Ningyuan Deng, Xinyuan Song, Ziqian Bi, Tianyang Wang, Zheyu Yao, Keyu Chen, Ming Li, Qian Niu, Junyu Liu, et al. Advanced deep learning methods for protein structure prediction and design.BIO Integration, 2025
2025
-
[3]
Highly accurate protein structure prediction with AlphaFold.Nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with AlphaFold.Nature, 596(7873):583–589, 2021
2021
-
[4]
R2i-bench: Benchmarking reasoning-driven text-to-image generation
Kaijie Chen, Zihao Lin, Zhiyang Xu, Ying Shen, Yuguang Yao, Joy Rimchala, Jiaxin Zhang, and Lifu Huang. R2i-bench: Benchmarking reasoning-driven text-to-image generation. In Proceedings of the 2025 Conference on Empirical Methods in Natural Language Processing, pages 12606–12641, 2025
2025
-
[5]
De novo design of protein structure and function with RFdiffusion.Nature, 620(7976):1089–1100, 2023
Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borber, Robert J Ragotte, Lukas F Milles, et al. De novo design of protein structure and function with RFdiffusion.Nature, 620(7976):1089–1100, 2023
2023
-
[6]
Illuminating protein space with a programmable generative model.Nature, 623(7989):1070–1078, 2023
John B Ingraham, Max Barber, Greta Wilber, Luke Strom, Chandra Theesfeld, Julia Listgarten, Gabriele Corso, Tommi Jaakkola, and Regina Barzilay. Illuminating protein space with a programmable generative model.Nature, 623(7989):1070–1078, 2023
2023
-
[7]
Protein generation with evolutionary diffusion: sequence is all you need
Sarah Alamdari, Nitya Thakkar, Rianne van den Berg, Alex X Lu, Nicolo Fusi, Ava P Amini, and Kevin K Yang. Protein generation with evolutionary diffusion: sequence is all you need. bioRxiv, pages 2023–09, 2023
2023
-
[8]
From noise to nuance: Advances in deep generative image models.arXiv:2412.09656, 2024
Benji Peng, Chia Xin Liang, Ziqian Bi, Ming Liu, Yichao Zhang, Tianyang Wang, Keyu Chen, Xinyuan Song, and Pohsun Feng. From noise to nuance: Advances in deep generative image models.arXiv:2412.09656, 2024
arXiv 2024
-
[9]
Drdgrl: Dual-relational dynamic graph repre- sentation learning for delay-sensitive stock trend prediction
Mingjie You, Kaijie Chen, and Dawei Cheng. Drdgrl: Dual-relational dynamic graph repre- sentation learning for delay-sensitive stock trend prediction. InInternational Conference on Database Systems for Advanced Applications, pages 35–50. Springer, 2026
2026
-
[10]
Haobo Zhang, Xutao Mao, Guangyuan Dong, Ziwei Li, Xuanbo Su, Kaijie Chen, Jing Yang, and Zheng Lin. Memmark: State-evolution attribution watermarking for agent long-term memory systems.arXiv preprint arXiv:2605.25002, 2026. 13
Pith/arXiv arXiv 2026
-
[11]
Denoising diffusion probabilistic models.Advances in Neural Information Processing Systems, 33:6840–6851, 2020
Jonathan Ho, Ajay Jain, and Pieter Abbeel. Denoising diffusion probabilistic models.Advances in Neural Information Processing Systems, 33:6840–6851, 2020
2020
-
[12]
Flow matching for generative modeling
Yaron Lipman, Ricky TQ Chen, Heli Ben-Hamu, Maximilian Nickel, and Matthew Le. Flow matching for generative modeling. InInternational Conference on Learning Representations, 2023
2023
-
[13]
Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
2023
-
[14]
Attention is all you need
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention is all you need. InAdvances in Neural Informa- tion Processing Systems, volume 30, 2017
2017
-
[15]
Prottrans: Toward understanding the language of life through self-supervised learning.IEEE Transactions on Pattern Analysis and Machine Intelligence, 44(10):7112–7127, 2022
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, et al. Prottrans: Toward understanding the language of life through self-supervised learning.IEEE Transactions on Pattern Analysis and Machine Intelligence, 44(10):7112–7127, 2022
2022
-
[16]
Structure-based protein function prediction using graph convolutional networks.Nature Communications, 12(1):3168, 2021
Vladimir Gligorijevi ´c, P Douglas Renfrew, Tomasz Kosciolek, Julia Koehler Ber, Daniel Berenberg, Tommi Vatez, Chris Chandler, Andre Taylor-Compston, Brendan J Frey, and Richard Bonneau. Structure-based protein function prediction using graph convolutional networks.Nature Communications, 12(1):3168, 2021
2021
-
[17]
Bowen Jing, Stephan Eismann, Patricia Suriana, Raphael JL Townshend, and Ron Dror. Equiv- ariant graph neural networks for 3d macromolecular structure.arXiv preprint arXiv:2106.03843, 2021
arXiv 2021
-
[18]
Generating functional protein variants with variational autoencoders.PLOS Computational Biology, 17(2):e1008736, 2021
Alex Hawkins-Hooker, Florence Depardieu, Adrian Baez-Ortega, Marie Touchon, Eduardo PC Rocha, Ilaria Granata, Michael PH Brown, and Michael A Savageau. Generating functional protein variants with variational autoencoders.PLOS Computational Biology, 17(2):e1008736, 2021
2021
-
[19]
Ig-vae: Generative modeling of protein structure by direct 3d coordinate generation.PLOS Computational Biology, 18(6):e1010271, 2022
Raphael R Eguchi, Christian A Choe, and Po-Ssu Huang. Ig-vae: Generative modeling of protein structure by direct 3d coordinate generation.PLOS Computational Biology, 18(6):e1010271, 2022
2022
-
[20]
Generative models for graph-based protein design
John Ingraham, Vikas Garg, Regina Barzilay, and Tommi Jaakkola. Generative models for graph-based protein design. InAdvances in Neural Information Processing Systems, volume 32, 2019
2019
-
[21]
Se(3) diffusion model with application to protein backbone generation
Jason Yim, Brian L Trippe, Valentin De Bortoli, Emile Mathieu, Arnaud Doucet, Regina Barzilay, and Tommi Jaakkola. Se(3) diffusion model with application to protein backbone generation. InInternational Conference on Machine Learning, pages 40001–40039. PMLR, 2023
2023
-
[22]
Huiyi Chen, Jiawei Peng, Dehai Min, Changchang Sun, Kaijie Chen, Yan Yan, Xu Yang, and Lu Cheng. Mvi-bench: A comprehensive benchmark for evaluating robustness to misleading visual inputs in lvlms.arXiv preprint arXiv:2511.14159, 2025
Pith/arXiv arXiv 2025
-
[23]
Gui agents for continual game generation.arXiv preprint arXiv:2605.28258, 2026
Yixu Huang, Bo Li, Na Li, Zhe Wang, Kaijie Chen, Haonan Ge, Qingyi Si, Yuanzhe Shen, Ruihan Yang, Guangjing Wang, et al. Gui agents for continual game generation.arXiv preprint arXiv:2605.28258, 2026
Pith/arXiv arXiv 2026
-
[24]
Superflow: Training flow matching models with rl on the fly.arXiv preprint arXiv:2512.17951, 2025
Kaijie Chen, Zhiyang Xu, Ying Shen, Zihao Lin, Yuguang Yao, and Lifu Huang. Superflow: Training flow matching models with rl on the fly.arXiv preprint arXiv:2512.17951, 2025. 14
arXiv 2025
-
[25]
Generating novel, designable, and diverse protein structures by equivariantly diffusing oriented residue clouds
Yeqing Lin and Mohammed AlQuraishi. Generating novel, designable, and diverse protein structures by equivariantly diffusing oriented residue clouds. InInternational Conference on Machine Learning, pages 21312–21333. PMLR, 2023
2023
-
[26]
Yeqing Lin, Minji Lee, Zhao Zhang, and Mohammed AlQuraishi. Out of many, one: Designing and scaffolding proteins at the scale of the structural universe with Genie 2.arXiv preprint arXiv:2405.15489, 2024
arXiv 2024
-
[27]
Se(3)-stochastic flow matching for protein backbone generation
Avishek Joey Bose, Tara Akhound-Sadegh, Kilian Fatras, Guillaume Huguet, Jarrid Rector- Brooks, Cheng-Hao Liu, Andrei Cristian Nica, Maksym Korablyov, Michael Bronstein, and Alexander Tong. Se(3)-stochastic flow matching for protein backbone generation. InInterna- tional Conference on Learning Representations, 2024
2024
-
[28]
Fast protein backbone generation with SE(3) flow matching.arXiv preprint arXiv:2310.05297, 2023
Jason Yim, Andrew Campbell, Andrew YK Foong, Michael Gastegger, José Jiménez-Luna, Sarah Lewis, Victor Garcia Satorras, Bastiaan S Veeling, Regina Barzilay, Tommi Jaakkola, et al. Fast protein backbone generation with SE(3) flow matching.arXiv preprint arXiv:2310.05297, 2023
arXiv 2023
-
[29]
Joint generation of protein sequence and structure with RoseTTAFold sequence space diffusion
Sidney Lyayuga Lisanza, Jake M Gershon, Sam WK Tipps, Jerald A Arnesen, Chenlin Zhu, Samuel J Zandberg, Rishi Raman, Casper Bakker, W Sebastian Koska, Dustin Lehnert, et al. Joint generation of protein sequence and structure with RoseTTAFold sequence space diffusion. bioRxiv, pages 2023–05, 2023
2023
-
[30]
Protgpt2 is a deep unsupervised language model for protein design.Nature Communications, 13(1):4348, 2022
Noelia Ferruz, Steffen Schmidt, and Birte Höcker. Protgpt2 is a deep unsupervised language model for protein design.Nature Communications, 13(1):4348, 2022
2022
-
[31]
Protein design with guided discrete diffusion.Advances in Neural Information Processing Systems, 36, 2024
Nate Gruver, Samuel Stanton, Nathan C Frey, Tim GJ Rudner, Isidro Hotzel, Julien Lafrance- Vanasse, Arvind Rajpal, Kyunghyun Cho, and Andrew Gordon Wilson. Protein design with guided discrete diffusion.Advances in Neural Information Processing Systems, 36, 2024
2024
-
[32]
E(n) equivariant graph neural networks
Víctor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E(n) equivariant graph neural networks. InInternational Conference on Machine Learning, pages 9323–9332. PMLR, 2021
2021
-
[33]
Se(3)-transformers: 3d roto-translation equivariant attention networks
Fabian Fuchs, Daniel Worrall, V olker Fischer, and Max Welling. Se(3)-transformers: 3d roto-translation equivariant attention networks. InAdvances in Neural Information Processing Systems, volume 33, pages 1970–1981, 2020
1970
-
[34]
Equiformer: Equivariant graph attention transformer for 3d atomistic graphs
Yi-Lun Liao and Tess Smidt. Equiformer: Equivariant graph attention transformer for 3d atomistic graphs. InInternational Conference on Learning Representations, 2023
2023
-
[35]
The protein data bank.Nucleic Acids Research, 28(1):235–242, 2000
Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. The protein data bank.Nucleic Acids Research, 28(1):235–242, 2000
2000
-
[36]
Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets.Nature Biotechnology, 35(11):1026–1028, 2017
Martin Steinegger and Johannes Söding. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets.Nature Biotechnology, 35(11):1026–1028, 2017
2017
-
[37]
Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models.Nucleic Acids Research, 50(D1):D419–D427, 2022
Mihaly Varadi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cyrus Natassia, Galabina Yordanova, David Yuan, Oana Stroe, Gemma Wood, Agata Laydon, et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models.Nucleic Acids Research, 50(D1):D419–D427, 2022
2022
-
[38]
Robust deep learning–based protein sequence design using ProteinMPNN.Science, 378(6615):49–56, 2022
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courber, Rob J de Haas, Neville Bethel, et al. Robust deep learning–based protein sequence design using ProteinMPNN.Science, 378(6615):49–56, 2022
2022
-
[39]
Diffdock: Diffusion steps, twists, and turns for molecular docking.International Conference on Learning Representations, 2023
Gabriele Corso, Hannes Stärk, Bowen Jing, Regina Barzilay, and Tommi Jaakkola. Diffdock: Diffusion steps, twists, and turns for molecular docking.International Conference on Learning Representations, 2023. 15
2023
-
[40]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, and Rob Fergus. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the National Academy of Sciences, 118(15):e2016239118, 2021
2021
-
[41]
Deepsol: a deep learning framework for sequence-based protein solubility prediction.Bioinformatics, 34(15):2605–2613, 2018
Sameer Khurana, Reda Rawi, Kumardeep Kuber, Shaomin Hadar, Ohad Manor, Christine Orengo, Douglas EV Pires, David B Ascher, Lenore Cowen, and Gaurav Bhardwaj. Deepsol: a deep learning framework for sequence-based protein solubility prediction.Bioinformatics, 34(15):2605–2613, 2018
2018
-
[42]
Openmm 7: Rapid development of high performance algorithms for molecular dynamics.PLOS Computational Biology, 13(7):e1005659, 2017
Peter Eastman, Jason Swails, John D Chodera, Robert T McGibbon, Yutong Zhao, Kyle A Beauchamp, Lee-Ping Wang, Andrew C Simmonett, Matthew P Harrigan, Chaya D Stern, et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics.PLOS Computational Biology, 13(7):e1005659, 2017
2017
-
[43]
Macromolecular modeling and design in Rosetta: recent methods and frameworks.Nature Methods, 17(7):665–680, 2020
Julia Koehler Leman, Brian D Weitzner, Steven M Lewis, Jared Adolf-Bryfogle, Nawsad Alam, Rebecca F Alford, Melanie Aprahamian, David Baker, Kyle A Barlow, Patrick Barth, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks.Nature Methods, 17(7):665–680, 2020
2020
-
[44]
Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic Acids Research, 33(7):2302–2309, 2005
Yang Zhang and Jeffrey Skolnick. Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic Acids Research, 33(7):2302–2309, 2005
2005
-
[45]
Neural ordinary differential equations
Ricky TQ Chen, Yulia Rubanova, Jesse Bettencourt, and David K Duvenaud. Neural ordinary differential equations. InAdvances in Neural Information Processing Systems, volume 31, 2018. 16 A Theoretical Analysis A.1 Convergence Guarantee for Hierarchical Flow Matching We provide theoretical justification for our hierarchical approach by analyzing the converge...
2018
-
[46]
Design 8 sequences using ProteinMPNN with sampling temperature 0.1 19 Table 6: Model hyperparameters. Parameter Stage 1 Stage 2 Hidden dimension 384 256 Number of layers 12 8 Attention heads 12 8 Dropout 0.1 0.1 Learning rate3×10 −4 3×10 −4 Batch size 256 128 Training steps 500K 300K
-
[47]
Predict structures for all sequences using ESMFold
-
[48]
Compute scTM score between generated backbone and predicted structures
-
[49]
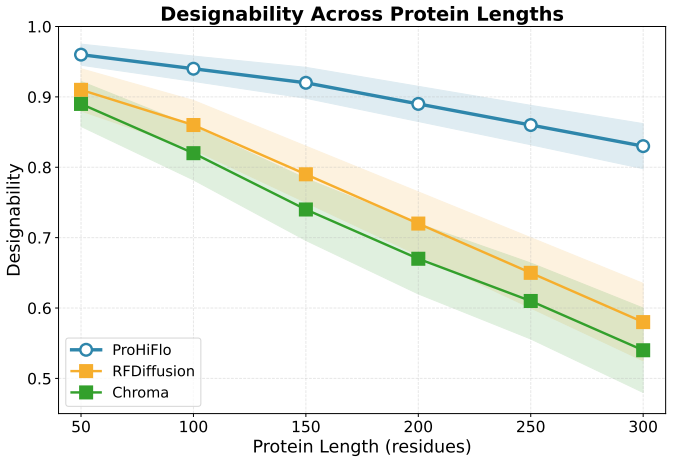
Report designability as fraction with max(scTM)>0.5 D Additional Results D.1 Per-Length Designability Breakdown Table 7: Designability breakdown by protein length. Method 50-100 100-150 150-200 200-250 250-300 RFDiffusion 0.912 0.867 0.798 0.723 0.651 Chroma 0.894 0.834 0.756 0.689 0.612 FoldFlow-2 0.923 0.889 0.834 0.778 0.712 ProHiFlo 0.967 0.945 0.912 ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.