Evidence-Aware Protein Complex Detection: Methods, Benchmarks, and Reproducibility Challenges
Pith reviewed 2026-06-28 08:12 UTC · model grok-4.3
The pith
Evidence-aware graph methods best balance plausibility and reproducibility in protein complex detection, but evaluation protocols are now the main limit.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
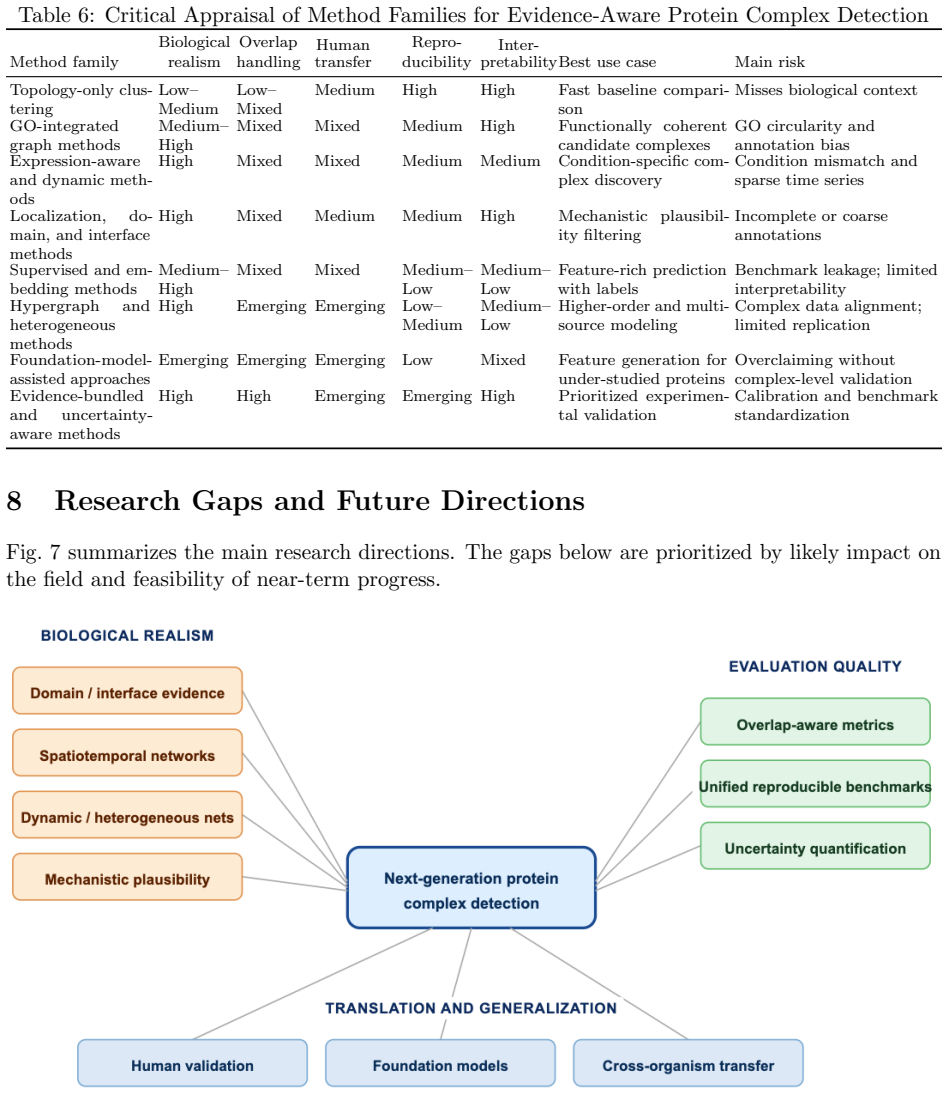
Transparent evidence-aware graph methods currently offer the strongest tradeoff between biological plausibility and reproducibility, while deep, hypergraph, and dynamic heterogeneous models expand biological realism but require stronger benchmark control. The central bottleneck is no longer only the lack of algorithms, but the lack of harmonized, overlap-aware, and reproducible evaluation protocols.
What carries the argument
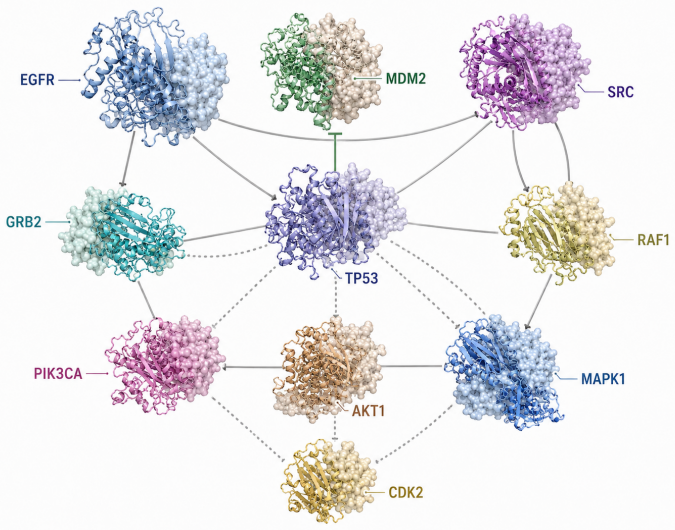
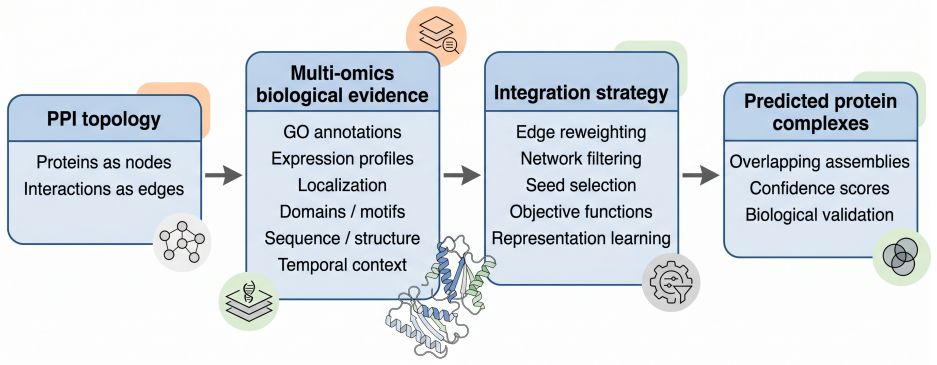
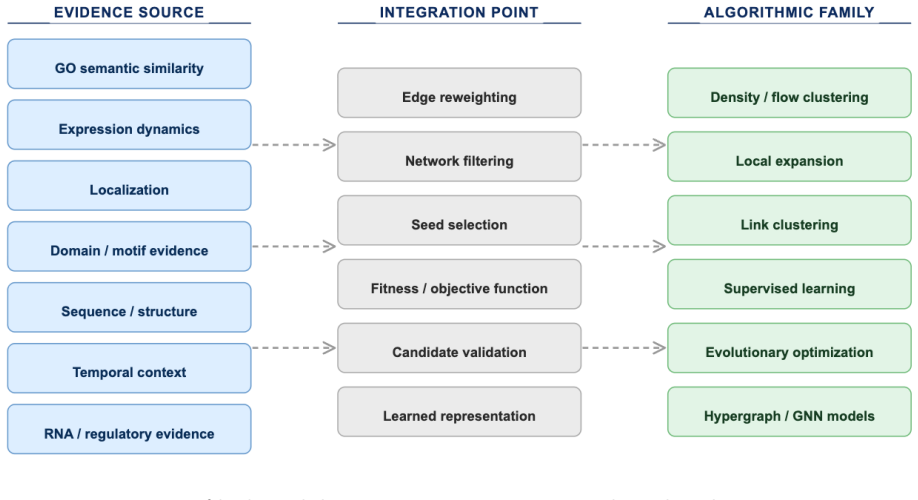
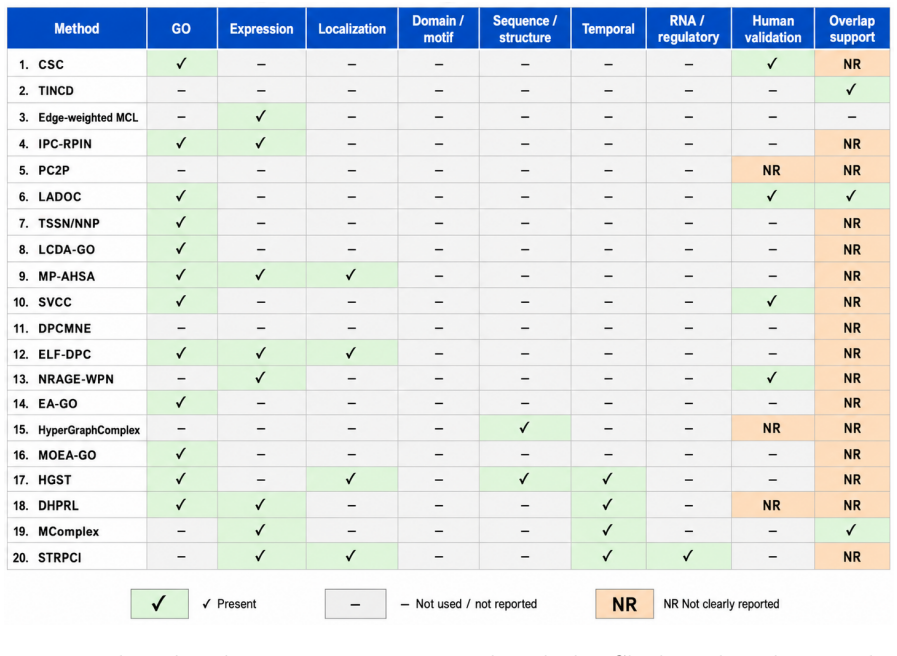
Evidence-aware approaches that combine PPI topology with Gene Ontology annotations, expression profiles, subcellular localization, and other supporting data sources.
If this is right
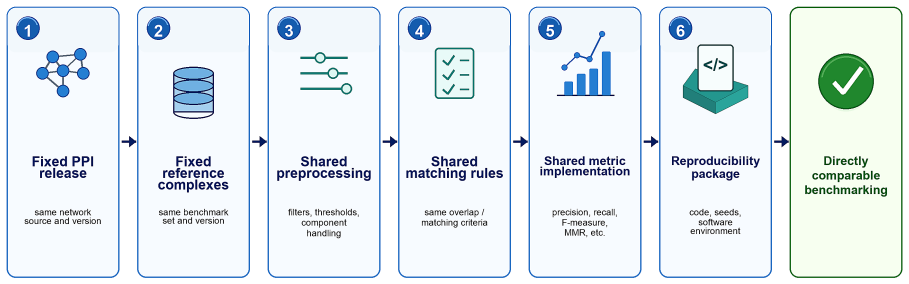
- Unified benchmark versions would enable direct comparison of methods without hidden differences in data processing.
- Explicit controls for circular use of Gene Ontology information would reduce inflated performance scores.
- Overlap-aware metrics would produce rankings that better match the biological reality of shared subunits in complexes.
- Routine reporting of uncertainty estimates would make performance claims more trustworthy across independent runs.
- Releasing executable software packages would allow other groups to verify and extend reported results.
Where Pith is reading between the lines
- Harmonized protocols developed here could be adapted to improve reproducibility in related tasks such as protein function prediction from networks.
- If evaluation standards tighten, researchers might test whether evidence-aware methods maintain their edge when applied to context-specific or tissue-specific interaction maps.
- The emphasis on transparent methods suggests that future work could prioritize interpretable models over black-box ones when integrating new data types like single-cell expression.
- Adopting the recommended controls might narrow the gap between computational predictions and what can be validated in targeted experiments.
Load-bearing premise
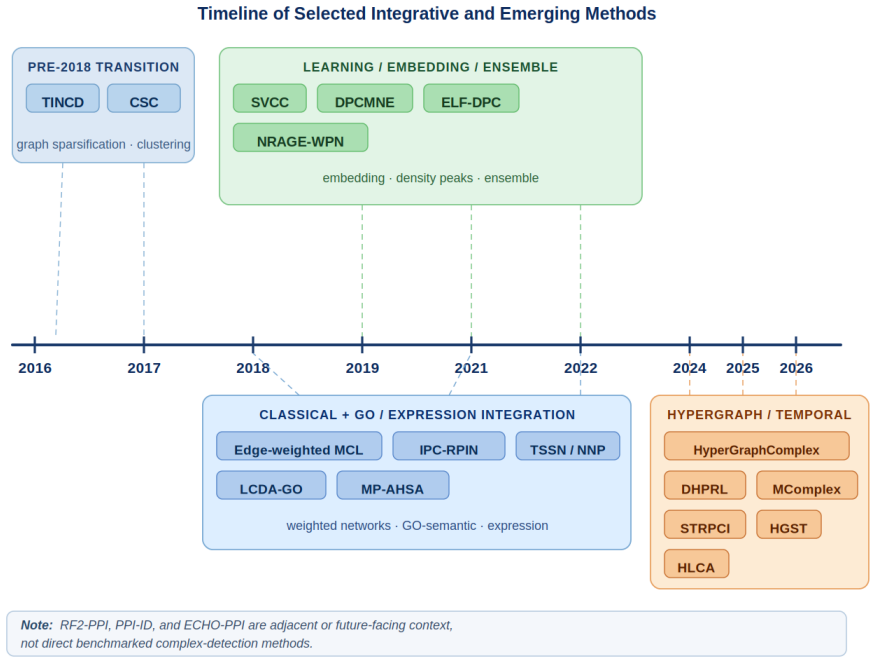
The post-2018 methods and selected historical baselines reviewed are representative enough of the literature to establish that evaluation protocols rather than new algorithms are the primary limiting factor.
What would settle it
A broader survey that includes many post-2018 methods omitted here and finds that non-evidence-aware or highly complex models achieve superior reproducible performance would undermine the tradeoff claim; implementing the recommended unified benchmarks and seeing no shift in which methods rank highest would falsify the bottleneck claim.
Figures
read the original abstract
Protein complexes are central units of cellular organization, yet their identification from protein-protein interaction (PPI) networks remains difficult because interactome maps are noisy, incomplete, context dependent, and unevenly annotated. This focused methodological review examines evidence-aware approaches that combine PPI topology with Gene Ontology (GO) annotations, expression profiles, subcellular localization, sequence or domain evidence, temporal information, and representation learning, with emphasis on post-2018 methods and selected historical baselines. The central synthesis is that transparent evidence-aware graph methods currently offer the strongest tradeoff between biological plausibility and reproducibility, while deep, hypergraph, and dynamic heterogeneous models expand biological realism but require stronger benchmark control. The central bottleneck is no longer only the lack of algorithms, but the lack of harmonized, overlap-aware, and reproducible evaluation protocols. We therefore recommend unified benchmark versions, explicit GO-circularity controls, overlap-aware metrics, uncertainty estimates, and executable software packages over isolated source-specific F-measure gains.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. This manuscript is a focused methodological review of evidence-aware protein complex detection from PPI networks. It surveys methods that integrate network topology with GO annotations, expression profiles, subcellular localization, sequence/domain evidence, temporal data, and representation learning, with particular attention to post-2018 approaches and selected earlier baselines. The central synthesis states that transparent evidence-aware graph methods currently provide the strongest tradeoff between biological plausibility and reproducibility, whereas deep, hypergraph, and dynamic heterogeneous models increase realism at the cost of requiring tighter benchmark controls. The paper concludes that the primary remaining bottleneck is the absence of harmonized, overlap-aware, and reproducible evaluation protocols, and therefore advocates unified benchmark versions, explicit GO-circularity controls, overlap-aware metrics, uncertainty estimates, and executable software packages.
Significance. If the synthesis holds, the review would be useful in redirecting community effort from isolated algorithmic novelty toward standardized, reproducible evaluation practices. The explicit recommendations for overlap-aware metrics and GO-circularity controls, together with the call for executable packages, constitute concrete, actionable guidance that could improve comparability across studies. The paper also usefully distinguishes the strengths of simpler evidence-aware graph methods from the added complexity of newer architectures.
major comments (1)
- [Abstract] Abstract: The claim that 'the central bottleneck is no longer only the lack of algorithms, but the lack of harmonized, overlap-aware, and reproducible evaluation protocols' is load-bearing for the central synthesis. This generalization requires that the reviewed post-2018 methods plus selected baselines are representative of the broader literature; without an explicit statement of literature search strategy, inclusion/exclusion criteria, or a systematic sampling frame (none provided in the abstract or visible in the synthesis), it remains possible that recent deep or hypergraph methods already incorporating overlap-aware or uncertainty-aware evaluation were omitted, which would undermine the conclusion that evaluation protocols are now the dominant issue.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. The major comment concerns the need for explicit documentation of literature selection to support the central claim in the abstract. We address this below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that 'the central bottleneck is no longer only the lack of algorithms, but the lack of harmonized, overlap-aware, and reproducible evaluation protocols' is load-bearing for the central synthesis. This generalization requires that the reviewed post-2018 methods plus selected baselines are representative of the broader literature; without an explicit statement of literature search strategy, inclusion/exclusion criteria, or a systematic sampling frame (none provided in the abstract or visible in the synthesis), it remains possible that recent deep or hypergraph methods already incorporating overlap-aware or uncertainty-aware evaluation were omitted, which would undermine the conclusion that evaluation protocols are now the dominant issue.
Authors: We acknowledge the referee's point. The manuscript is presented as a focused methodological review of evidence-aware methods (explicitly combining PPI topology with GO, expression, localization, sequence/domain, temporal, or representation-learning evidence), not a systematic review. Selection was guided by coverage of post-2018 approaches meeting these criteria plus key baselines, drawn from recent surveys and field knowledge. To strengthen the paper, we will add an explicit 'Scope and Selection Criteria' paragraph (in the Introduction or a new subsection) describing the search strategy (PubMed, arXiv, Google Scholar; keywords combining 'protein complex detection', 'PPI network', 'evidence integration' or specific evidence types; post-2018 filter; inclusion of methods reporting benchmark performance). This will clarify the scope without altering the synthesis for the reviewed class of methods. We maintain that the central claim holds within this focused scope but agree explicit documentation is warranted. revision: yes
Circularity Check
Literature synthesis review with no derivation chain or self-referential reductions
full rationale
This is a methodological review paper synthesizing existing literature on protein complex detection methods, with emphasis on evidence-aware approaches and evaluation protocols. No equations, fitted parameters, predictions, or mathematical derivations are present that could reduce to inputs by construction. The central synthesis—that evaluation protocols are now the primary bottleneck—rests on a survey of post-2018 methods and baselines rather than any self-definitional, fitted-input, or self-citation load-bearing step. Representativeness of the selected methods is a sampling/validity concern external to the paper's internal logic, not a circularity pattern. The paper is self-contained as a literature synthesis against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
A comprehensive two-hybrid analysis to explore the yeast protein interactome,
T. Itoet al., “A comprehensive two-hybrid analysis to explore the yeast protein interactome,” Proc. Natl. Acad. Sci. USA, vol. 98, no. 8, pp. 4569–4574, 2001, doi: 10.1073/pnas.061034498
-
[2]
A. Bauer and B. Kuster, “Affinity purification-mass spectrometry: powerful tools for the characterization of protein complexes,”Eur. J. Biochem., vol. 270, no. 4, pp. 570–578, 2003, doi: 10.1046/j.1432-1033.2003.03424.x
-
[3]
Protein complex prediction: A survey,
J. Zahiriet al., “Protein complex prediction: A survey,”Genomics, vol. 112, no. 1, pp. 174–183, 2020, doi: 10.1016/j.ygeno.2019.01.011
-
[4]
A survey of computational methods for protein complex prediction from protein interaction networks,
S. Srihari and H. W. Leong, “A survey of computational methods for protein complex prediction from protein interaction networks,”J. Bioinform. Comput. Biol., vol. 11, no. 2, 1230002, 2013
2013
-
[5]
Methods for protein complex prediction and their contributions towards understanding the organisation, function and dynamics of complexes,
S. Srihariet al., “Methods for protein complex prediction and their contributions towards understanding the organisation, function and dynamics of complexes,”FEBS Lett., vol. 589, no. 19, pp. 2590–2602, 2015
2015
-
[6]
Computational approaches for detecting protein complexes from protein interaction networks: a survey,
X. Li, M. Wu, C. K. Kwohet al., “Computational approaches for detecting protein complexes from protein interaction networks: a survey,”BMC Genomics, vol. 11, Suppl. 1, S3, 2010
2010
-
[7]
G. D. Bader and C. W. V. Hogue, “An automated method for finding molecular complexes in large protein interaction networks,”BMC Bioinformatics, vol. 4, 2, 2003, doi: 10.1186/1471- 2105-4-2. 19
-
[8]
An efficient algorithm for large-scale detection of protein families,
A. J. Enright, S. Van Dongen, and C. A. Ouzounis, “An efficient algorithm for large-scale detection of protein families,”Nucleic Acids Res., vol. 30, no. 7, pp. 1575–1584, 2002
2002
-
[9]
CFinder: locating cliques and overlapping modules in biological networks,
B. Adamcsek, G. Palla, I. J. Farkaset al., “CFinder: locating cliques and overlapping modules in biological networks,”Bioinformatics, vol. 22, no. 8, pp. 1021–1023, 2006
2006
-
[10]
Detecting overlapping protein complexes in protein- protein interaction networks,
T. Nepusz, H. Yu, and A. Paccanaro, “Detecting overlapping protein complexes in protein- protein interaction networks,”Nat. Methods, vol. 9, no. 5, pp. 471–472, 2012, doi: 10.1038/nmeth.1938
-
[11]
Protein complex prediction via cost-based clustering,
A. D. King, N. Przulj, and I. Jurisica, “Protein complex prediction via cost-based clustering,” Bioinformatics, vol. 20, no. 17, pp. 3013–3020, 2004
2004
-
[12]
Discovery of protein complexes with core-attachment structures from tandem affinity purification data,
M. Wu, X. Li, C. K. Kwohet al., “Discovery of protein complexes with core-attachment structures from tandem affinity purification data,”J. Comput. Biol., vol. 19, no. 9, pp. 1027– 1042, 2012
2012
-
[13]
Performance evaluation measures for protein complex prediction,
A. Ivazehet al., “Performance evaluation measures for protein complex prediction,”Genomics, vol. 111, no. 6, pp. 1483–1492, 2019
2019
-
[14]
Topological and functional comparison of community detection algorithms in biological networks,
S. Rahiminejad, M. R. Maurya, and S. Subramaniam, “Topological and functional comparison of community detection algorithms in biological networks,”BMC Bioinformatics, vol. 20, 212, 2019
2019
-
[15]
An improved method for scoring protein-protein interactions using semantic similarity within the Gene Ontology,
S. Jain and G. D. Bader, “An improved method for scoring protein-protein interactions using semantic similarity within the Gene Ontology,”BMC Bioinformatics, vol. 11, 562, 2010
2010
-
[16]
The Gene Ontology resource: enriching a GOld mine,
The Gene Ontology Consortium, “The Gene Ontology resource: enriching a GOld mine,” Nucleic Acids Res., vol. 49, no. D1, pp. D325–D334, 2021
2021
-
[17]
P. Sharma, D. K. Bhattacharyya, and J. K. Kalita, “Detecting protein complexes based on a combination of topological and biological properties in protein-protein interaction network,”J. Genet. Eng. Biotechnol., vol. 16, no. 1, pp. 217–226, 2018, doi: 10.1016/j.jgeb.2017.11.005
-
[18]
Detecting complexes from edge-weighted PPI networks via genes expression analysis,
Z. Zhang, J. Song, J. Tanget al., “Detecting complexes from edge-weighted PPI networks via genes expression analysis,”BMC Syst. Biol., vol. 12, Suppl. 4, 40, 2018, doi: 10.1186/s12918- 018-0565-y
-
[19]
W. Zhang, J. Xu, Y. Liet al., “Integrating network topology, gene expression data and GO annotation information for protein complex prediction,”J. Bioinform. Comput. Biol., vol. 17, no. 1, 1950001, 2019, doi: 10.1142/S021972001950001X
-
[20]
An effective link-based clustering algorithm for detecting overlapping protein complexes in protein-protein interaction networks,
L. Huet al., “An effective link-based clustering algorithm for detecting overlapping protein complexes in protein-protein interaction networks,”IEEE Trans. Netw. Sci. Eng., vol. 8, no. 4, pp. 3275–3289, 2021
2021
-
[21]
A new method for recognizing protein complexes based on protein interaction networks and GO terms,
X. Wang, N. Zhang, Y. Zhaoet al., “A new method for recognizing protein complexes based on protein interaction networks and GO terms,”Front. Genet., vol. 12, 792265, 2021, doi: 10.3389/fgene.2021.792265
-
[22]
From communities to protein complexes: A local community detection algorithm on PPI networks,
S. Dilmaghani, M. R. Brust, C. H. C. Ribeiroet al., “From communities to protein complexes: A local community detection algorithm on PPI networks,”PLoS ONE, vol. 17, no. 1, e0260484, 2022, doi: 10.1371/journal.pone.0260484. 20
-
[23]
Detecting protein complexes with multiple properties by an adaptive harmony search algorithm,
R. Wang, C. Wang, and H. Ma, “Detecting protein complexes with multiple properties by an adaptive harmony search algorithm,”BMC Bioinformatics, vol. 23, 414, 2022, doi: 10.1186/s12859-022-04923-4
-
[24]
X. Wanget al., “A supervised protein complex prediction method with network representa- tion learning and gene ontology knowledge,”BMC Bioinformatics, vol. 23, 300, 2022, doi: 10.1186/s12859-022-04850-4
-
[25]
Y. Yu and D. Kong, “Protein complexes detection based on node local properties and gene ex- pression in PPI weighted networks,”BMC Bioinformatics, vol. 23, 24, 2022, doi: 10.1186/s12859- 021-04543-4
-
[26]
S. A. Abdulsahib and B. A. Attea, “An evolutionary algorithm for improving the quantity and quality of the detected complexes from protein interaction networks,”Iraqi J. Sci., vol. 65, no. 5, pp. 2898–2924, 2024, doi: 10.24996/ijs.2024.65.5.42
-
[27]
M. N. Abbas, D. Broneske, and G. Saake, “A multi-objective evolutionary algorithm for detecting protein complexes in PPI networks using gene ontology,”Sci. Rep., vol. 15, 16855, 2025, doi: 10.1038/s41598-025-01667-y
-
[28]
S. Wang, H. Cui, Y. Quet al., “Multi-source biological knowledge-guided hypergraph spa- tiotemporal subnetwork embedding for protein complex identification,”Brief. Bioinform., vol. 26, no. 1, bbae718, 2025, doi: 10.1093/bib/bbae718
-
[29]
Protein complex prediction via verifying and reconstructing the topology of domain-domain interactions,
Y. Ozawaet al., “Protein complex prediction via verifying and reconstructing the topology of domain-domain interactions,”BMC Bioinformatics, vol. 11, 350, 2010
2010
-
[30]
Protein complex prediction based on maximum matching with domain-domain interaction,
W. Maet al., “Protein complex prediction based on maximum matching with domain-domain interaction,”Biochim. Biophys. Acta Proteins Proteom., vol. 1824, no. 12, pp. 1418–1424, 2012
2012
-
[31]
Identifying protein complexes based on an edge weight algorithm and core-attachment structure,
R. Wang, G. Liu, and C. Wang, “Identifying protein complexes based on an edge weight algorithm and core-attachment structure,”BMC Bioinformatics, vol. 20, 471, 2019, doi: 10.1186/s12859-019-3007-y
-
[32]
Recent advances in deep learning for protein-protein interaction analysis: A compre- hensive review,
M. Lee, “Recent advances in deep learning for protein-protein interaction analysis: A compre- hensive review,”Molecules, vol. 28, no. 13, 5169, 2023, doi: 10.3390/molecules28135169
-
[33]
X. Meng, J. Xiang, R. Zhenget al., “DPCMNE: Detecting protein complexes from protein- protein interaction networks via multi-level network embedding,”IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 19, no. 3, pp. 1592–1602, 2022, doi: 10.1109/TCBB.2021.3050102
-
[34]
PC2P: parameter-free network-based prediction of protein complexes,
S. Omranian, A. Angeleska, and Z. Nikoloski, “PC2P: parameter-free network-based prediction of protein complexes,”Bioinformatics, vol. 37, no. 1, pp. 73–81, 2021, doi: 10.1093/bioinfor- matics/btaa1089
-
[35]
DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions,
I. Xenarioset al., “DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions,”Nucleic Acids Res., vol. 30, no. 1, pp. 303–305, 2002
2002
-
[36]
Development of Human Protein Reference Database as an initial platform for approaching systems biology in humans,
S. Periet al., “Development of Human Protein Reference Database as an initial platform for approaching systems biology in humans,”Genome Res., vol. 13, no. 10, pp. 2363–2371, 2003
2003
-
[37]
MIPS: analysis and annotation of proteins from whole genomes,
H. W. Meweset al., “MIPS: analysis and annotation of proteins from whole genomes,”Nucleic Acids Res., vol. 32, suppl. 1, pp. D41–D44, 2004. 21
2004
-
[38]
Up-to-date catalogues of yeast protein complexes,
S. Pu, J. Wong, B. Turneret al., “Up-to-date catalogues of yeast protein complexes,”Nucleic Acids Res., vol. 37, no. 3, pp. 825–831, 2009
2009
-
[39]
PCDq: human protein complex database with quality index,
S. Kikugawaet al., “PCDq: human protein complex database with quality index,”BMC Syst. Biol., vol. 6, Suppl. 2, S7, 2012
2012
-
[40]
CORUM: the comprehensive resource of mammalian protein complexes–2019,
M. Giurgiuet al., “CORUM: the comprehensive resource of mammalian protein complexes–2019,” Nucleic Acids Res., vol. 47, no. D1, pp. D559–D563, 2019
2019
-
[41]
STRING v10: protein-protein interaction networks, integrated over the tree of life,
D. Szklarczyket al., “STRING v10: protein-protein interaction networks, integrated over the tree of life,”Nucleic Acids Res., vol. 43, no. D1, pp. D447–D452, 2015
2015
-
[42]
Human Protein Reference Database–2009 update,
T. S. Keshava Prasadet al., “Human Protein Reference Database–2009 update,”Nucleic Acids Res., vol. 37, suppl. 1, pp. D767–D772, 2009
2009
-
[43]
Objective criteria for the evaluation of clustering methods,
W. M. Rand, “Objective criteria for the evaluation of clustering methods,”J. Amer. Stat. Assoc., vol. 66, no. 336, pp. 846–850, 1971
1971
-
[44]
Comparing partitions,
L. Hubert and P. Arabie, “Comparing partitions,”J. Classification, vol. 2, pp. 193–218, 1985
1985
-
[45]
Using diversity in cluster ensembles,
L. I. Kuncheva and S. T. Hadjitodorov, “Using diversity in cluster ensembles,” inProc. IEEE Int. Conf. Syst., Man Cybern., 2004, vol. 2, pp. 1214–1219
2004
-
[46]
Detecting protein complexes in protein interaction networks modeled as gene expression biclusters,
E. M. Hanna, N. Zaki, and A. Amin, “Detecting protein complexes in protein interaction networks modeled as gene expression biclusters,”PLoS ONE, vol. 10, no. 12, e0144163, 2015, doi: 10.1371/journal.pone.0144163
-
[47]
PPI-ID: Streamlining protein-protein interaction prediction through domain and SLiM mapping,
H. V. Goodwin and N. S. Atkinson, “PPI-ID: Streamlining protein-protein interaction prediction through domain and SLiM mapping,”PLOS Computational Biology, vol. 21, no. 10, e1013062, 2025, doi: 10.1371/journal.pcbi.1013062
-
[48]
Predicting protein-protein interactions in the human proteome,
J. Zhanget al., “Predicting protein-protein interactions in the human proteome,”Science, vol. 390, no. 6771, eadt1630, 2025, doi: 10.1126/science.adt1630
-
[49]
Scientific Reports14(1), 23053 (2024)
M. Rayka and S. S. Naghavi, “Uncertainty quantification enables reliable deep learning for protein–ligand binding affinity prediction,”Sci. Rep., vol. 15, 43156, 2025, doi: 10.1038/s41598- 025-27167-7
-
[50]
An ensemble learning framework for detecting protein com- plexes from PPI networks,
R. Wang, H. Ma, and C. Wang, “An ensemble learning framework for detecting protein com- plexes from PPI networks,”Front. Genet., vol. 13, 839949, 2022, doi: 10.3389/fgene.2022.839949
-
[51]
S. Xia, D. Li, X. Denget al., “Integration of protein sequence and protein-protein interaction data by hypergraph learning to identify novel protein complexes,”Brief. Bioinform., vol. 25, no. 4, bbae274, 2024, doi: 10.1093/bib/bbae274
-
[52]
A two-layer integration framework for protein complex detection,
L. Ou-Yang, M. Wu, X.-F. Zhanget al., “A two-layer integration framework for protein complex detection,”BMC Bioinformatics, vol. 17, 100, 2016, doi: 10.1186/s12859-016-0939-3
-
[53]
S. Omranian, Z. Nikoloski, and D. G. Grimm, “Computational identification of protein complexes from network interactions: Present state, challenges, and the way forward,”Comput. Struct. Biotechnol. J., vol. 20, pp. 2699–2712, 2022, doi: 10.1016/j.csbj.2022.05.049. 22
-
[54]
S. Soltani, M. Jalali, and Y. Forghani, “ECHO-PPI: Trustworthy AI for evidence-bundled detection of overlapping protein modules in protein-protein interaction networks,”arXiv preprint arXiv:2605.21216, 2026, doi: 10.48550/arXiv.2605.21216
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.21216 2026
-
[55]
Z. Li, Y. Zhang, and P. Zhou, “Temporal protein complex identification based on dynamic het- erogeneous protein information network representation learning,”IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 21, no. 5, pp. 1154–1164, 2024, doi: 10.1109/TCBB.2024.3351078
-
[56]
L. Daou and E. M. Hanna, “Predicting protein complexes in protein interaction networks using Mapper and graph convolution networks,”Comput. Struct. Biotechnol. J., vol. 23, pp. 3595–3609, 2024, doi: 10.1016/j.csbj.2024.10.009
-
[57]
Spatiotemporal constrained RNA-protein heterogeneous network for protein complex identification,
Z. Li, S. Wang, H. Cuiet al., “Spatiotemporal constrained RNA-protein heterogeneous network for protein complex identification,”Brief. Bioinform., vol. 25, no. 4, bbae280, 2024, doi: 10.1093/bib/bbae280
-
[58]
J. Wang, X. Yang, P. Yanget al., “A graph clustering algorithm with hypergraph learning and a core-attachment strategy for protein complex identification,”Front. Genet., vol. 17, 1770432, 2026, doi: 10.3389/fgene.2026.1770432
-
[59]
Protein complex identification based on heterogeneous protein information network,
P. Zhou, Y. Zhang, Z. Liet al., “Protein complex identification based on heterogeneous protein information network,”J. Comput. Biol., vol. 30, no. 9, pp. 985–998, 2023, doi: 10.1089/cmb.2023.0081. 23
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.