Quantum Monte Carlo description of correlated electrons in two-dimensional FeSe
Pith reviewed 2026-05-22 16:33 UTC · model grok-4.3
The pith
Correlation energy in double-layer FeSe is set mostly by its atoms rather than by bonds between them.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
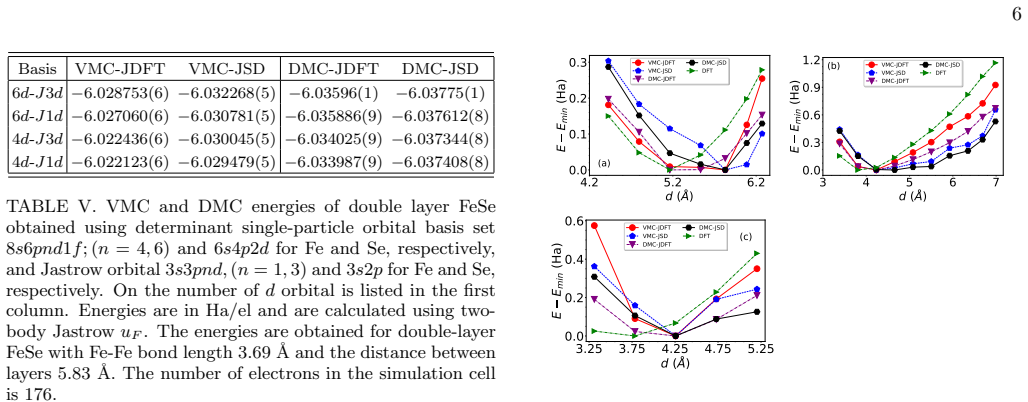
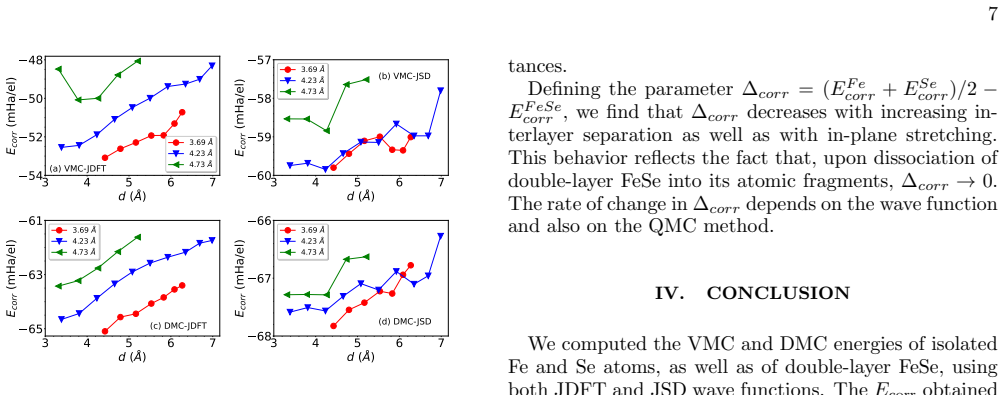
Variational and diffusion quantum Monte Carlo calculations with two forms of Slater-Jastrow trial wave functions show that the correlation energy of double-layer FeSe at the thermodynamic limit is dominated by the atomic contributions; the bonding between atoms plays only a minor role. When the interlayer separation is optimized under tensile strain, the correlation energy approaches that of the constituent Fe and Se atoms as stretch and spacing increase.
What carries the argument
Direct comparison of quantum Monte Carlo correlation energies between double-layer FeSe configurations and isolated Fe and Se atoms, extrapolated to the thermodynamic limit.
If this is right
- The correlation energy of double-layer FeSe moves closer to the atomic value as tensile strain and interlayer spacing grow.
- Atomic contributions outweigh bonding effects in setting the total correlation energy for the structures examined.
- Different geometrical configurations change the correlation energy but keep it close to the sum of atomic values.
Where Pith is reading between the lines
- Similar atomic dominance might appear in other layered transition-metal compounds and could be checked by the same method.
- Models that treat correlation largely at the atomic level may give useful estimates for correlation-driven properties in these materials.
- The approach could be extended to single-layer FeSe or to doped variants to test whether the atomic picture survives changes in dimensionality or carrier density.
Load-bearing premise
The Slater-Jastrow trial wave functions and the extrapolation to the thermodynamic limit give unbiased estimates of correlation-energy differences between the double-layer system and the separate atoms.
What would settle it
A calculation with a substantially different trial wave function or a direct experimental energy measurement that finds a large bonding contribution to the correlation energy would falsify the claim.
Figures




read the original abstract
An interesting question in physics is how the correlation energy of atoms evolves upon forming a solid. Here, we address this problem for a specific case of double-layer FeSe. We used many-body wavefunction-based quantum Monte Carlo (QMC) techniques to compute the correlation energies of double-layer FeSe with different geometrical configurations and compared them with those of isolated Fe and Se atoms. Variational and diffusion QMC calculations were carried out with Slater Jastrow trial wavefunctions employing two alternative forms for the homogeneous two-body pair correlation term. The ground-state energy was obtained in the thermodynamic limit using two types of trial wave functions of JDFT, in which only the Jastrow factor is optimized while the Slater determinant is derived from the local density approximation, and JSD, where both the Jastrow factor and the Slater determinant are optimized simultaneously. Our results indicate that the correlation energy of double layer FeSe at the thermodynamic limit is mainly determined by the atomic contributions, with the bonding between atoms playing a comparatively minor role in it. After optimizing the interlayer separation of double-layer FeSe under tensile strain, we analyze the correlation energy as a function of strain and separation. We found that with increasing tensile stretch and interlayer spacing, the correlation energy of double-layer FeSe stochastically approaches that of its constituent atomic fragments.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript applies variational Monte Carlo (VMC) and diffusion Monte Carlo (DMC) with Slater-Jastrow trial wavefunctions (JDFT and JSD forms) to compute the correlation energy of double-layer FeSe. Energies are extrapolated to the thermodynamic limit and compared directly to those of isolated Fe and Se atoms. The central claim is that atomic contributions dominate the correlation energy, with interatomic bonding playing a minor role; this is further examined under tensile strain and varying interlayer separation, where the layer correlation energy is reported to approach the atomic value with increasing stretch and spacing.
Significance. If the fixed-node errors cancel as assumed, the work supplies a first-principles QMC benchmark showing that correlation in this 2D FeSe system is largely atomic rather than bonding-driven. This could constrain effective models of its electronic structure and help interpret its superconductivity. The explicit use of two trial-wavefunction forms and thermodynamic-limit extrapolation constitutes a concrete strength.
major comments (2)
- [§3] §3 (DMC section) and the thermodynamic-limit extrapolation paragraph: the manuscript does not demonstrate or quantify cancellation of fixed-node errors between the periodic double-layer system (whose nodes reflect band dispersion) and the isolated atoms (whose nodes reflect multiplet structure). Because the central claim rests on the solid-minus-atoms correlation-energy difference being physically meaningful rather than an artifact of unequal fixed-node penalties, this omission is load-bearing.
- [Results on strain] Results on strain and interlayer separation: the reported stochastic approach of the layer correlation energy to the atomic value with increasing tensile stretch is presented without error bars on the difference or a test that residual finite-size and fixed-node contributions remain smaller than the observed trend. This weakens the quantitative support for the 'minor role of bonding' conclusion.
minor comments (2)
- [Abstract] The abstract states the main conclusion but contains no numerical values, error estimates, or explicit comparison numbers; adding at least one representative energy difference (with uncertainty) would improve readability.
- [Methods] Notation for the two-body Jastrow term is introduced without an explicit functional form or reference to the precise parametrization used in the JDFT versus JSD cases.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. We address the major points raised below and agree that additional discussion and analysis will strengthen the presentation of our results.
read point-by-point responses
-
Referee: [§3] §3 (DMC section) and the thermodynamic-limit extrapolation paragraph: the manuscript does not demonstrate or quantify cancellation of fixed-node errors between the periodic double-layer system (whose nodes reflect band dispersion) and the isolated atoms (whose nodes reflect multiplet structure). Because the central claim rests on the solid-minus-atoms correlation-energy difference being physically meaningful rather than an artifact of unequal fixed-node penalties, this omission is load-bearing.
Authors: We acknowledge that the manuscript does not provide a quantitative estimate of residual fixed-node errors in the solid-atom energy difference. Our calculations use the same Slater-Jastrow forms (JDFT and JSD) for both the double-layer FeSe and the isolated atoms, and the consistency of results between these two trial wavefunctions indicates that differences in nodal surfaces do not drive the main trends. We will revise the manuscript to include an expanded discussion of the fixed-node approximation, the rationale for expecting substantial cancellation in energy differences when using consistent trial functions, and the limitations of this assumption. A rigorous numerical quantification of the cancellation, however, would require methods such as release-node DMC that are beyond the scope of the present study. revision: yes
-
Referee: [Results on strain] Results on strain and interlayer separation: the reported stochastic approach of the layer correlation energy to the atomic value with increasing tensile stretch is presented without error bars on the difference or a test that residual finite-size and fixed-node contributions remain smaller than the observed trend. This weakens the quantitative support for the 'minor role of bonding' conclusion.
Authors: We agree that explicit error bars on the differences and additional checks would improve the quantitative support. In the revised manuscript we will report statistical uncertainties on the correlation-energy differences, include propagated error bars in the relevant figures, and add a brief analysis comparing results across different supercell sizes and the two trial wavefunctions to confirm that finite-size and fixed-node residuals are smaller than the observed trends with strain and interlayer spacing. revision: yes
- A complete numerical quantification of residual fixed-node errors in the solid-minus-atoms correlation energy difference.
Circularity Check
Direct first-principles QMC comparison of solid and atomic correlation energies shows no circularity
full rationale
The paper computes total energies via variational and diffusion Monte Carlo using Slater-Jastrow trial functions (JDFT with LDA-derived determinant and JSD with variationally optimized determinant) for both the double-layer FeSe supercells and the isolated Fe/Se atoms. Correlation energies are obtained from these total energies, extrapolated to the thermodynamic limit via finite-size scaling for the periodic system, and then compared numerically to quantify the difference attributable to bonding. This comparison is a direct subtraction of independently calculated quantities with no fitted parameters, no self-referential definitions of the target observable, and no load-bearing self-citations that reduce the central claim to prior inputs. The procedure is self-contained and externally falsifiable by other many-body methods.
Axiom & Free-Parameter Ledger
free parameters (1)
- Jastrow factor variational parameters
axioms (1)
- domain assumption Slater determinant derived from LDA or jointly optimized suffices to capture essential correlations
Reference graph
Works this paper leans on
-
[1]
basis set. The core electrons of the Fe and Se atoms were replaced by correlation consistent effective core po- tentials (ccECPs)[47, 48]. Our simulation cell is subject to two-dimensional periodic boundary conditions in the xydirection with standard Ewald summations and the Γ-point to calculate the Coulomb interaction. The varia- tional parameters in our...
-
[2]
andu C = b 2(1−exp(−r/b)) [52], whereris the relative distance between two electrons,aandbare vari- ational parameters. For Jastrow single-particle orbitals, we used uncontracted Gaussian basis sets of 3s3p1dand 3s2pfor Fe and Se, respectively. The DMC energies are obtained using the time step 0.01 a.u., the 3200 walkers (configurations), and the locality...
work page 2023
-
[3]
Q. H. Wang, A. Bedoya-Pinto, M. Blei, A. H. Dismukes, A. Hamo, S. Jenkins, M. Koperski, Y. Liu, Q.-C. Sun, E. J. Telford, H. H. Kim, M. Augustin, U. Vool, J.- X. Yin, L. H. Li, A. Falin, C. R. Dean, F. Casanova, R. F. Evans, M. Chshiev, A. Mishchenko, C. Petrovic, R. He, L. Zhao, A. W. Tsen, B. D. Gerardot, M. Brotons- Gisbert, Z. Guguchia, X. Roy, S. Ton...
work page 2022
-
[4]
M. Gibertini, M. Koperski, A. Morpurgo, and K. Novoselov, Magnetic 2d materials and heterostruc- tures, Nature nanotechnology14, 408 (2019)
work page 2019
-
[5]
S. Jenkins, L. R´ ozsa, U. Atxitia, R. F. L. Evans, K. S. Novoselov, and E. J. G. Santos, Breaking through the mermin-wagner limit in 2D van der waals magnets, Nat. Comm.13, 6917 (2022)
work page 2022
-
[6]
C. Gong and et al, Discovery of intrinsic ferromagnetism in two dimensional van der waals crystals, Nature546, 265 (2017)
work page 2017
-
[7]
H. Kontani and S. Onari, Orbital-fluctuation-mediated superconductivity in iron pnictides: Analysis of the five- orbital hubbard-holstein model, Phys. Rev. Lett.104, 157001 (2010)
work page 2010
-
[8]
A. E. B¨ ohmer and A. Kreisel, Nematicity, magnetism and superconductivity in FeSe, J. Phys.: Condensed Matter 30, 023001 (2018)
work page 2018
-
[9]
S. Medvedev, T. M. McQueen, I. A. Troyan, T. Palasyuk, M. I. Eremets, R. J. Cava, S. Naghavi, F. Casper, V. Ksenofontov, G. Wortmann, and C. Felser, Electronic and magnetic phase diagram ofβ-Fe 1.01Se with super- conductivity at 36.7 K under pressure, Nat. Mater.8, 630 (2009)
work page 2009
-
[10]
B. Kang, M. Kim, C. H. Park, and A. Janotti, Mott- insulator state of FeSe as a van der waals 2D material is unveiled, Phys. Rev. Lett.132, 266506 (2024)
work page 2024
-
[11]
Dagotto, Colloquium: The unexpected properties of alkali metal iron selenide superconductors, Rev
E. Dagotto, Colloquium: The unexpected properties of alkali metal iron selenide superconductors, Rev. Mod. Phys.85, 849 (2013)
work page 2013
-
[12]
Dai, Antiferromagnetic order and spin dynamics in iron-based superconductors, Rev
P. Dai, Antiferromagnetic order and spin dynamics in iron-based superconductors, Rev. Mod. Phys.87, 855 (2015)
work page 2015
- [13]
-
[14]
K. Kothapalli, A. B¨ ohmer, W. Jayasekara, B. Ueland, P. Das, A. S. V. Taufour, Y. Xiao, E. Alp, S. Bud’ko, P. Canfield, A. Kreyssig, and A. Goldman, Strong co- operative coupling of pressure-induced magnetic order and nematicity in FeSe, Nat. Commun. 7:127287, 12728 (2016)
work page 2016
-
[15]
B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo- Herrero, and X. Xu, Layer-dependent ferromagnetism in a van der waals crystal down to the monolayer limit, Nature546, 270 (2017)
work page 2017
- [16]
-
[17]
L. D. Landau, On the theory of the fermi liquid, Sov. Phys. JETP35, 95 (1958)
work page 1958
- [18]
-
[19]
A. V. Chubukov, M. Khodas, and R. M. Fernandes, Su- perconductivity, and spontaneous orbital order in iron- based superconductors: Which comes first and why?, Phys. Rev. X6, 041045 (2016)
work page 2016
-
[20]
Y. Yamakawa, S. Onari, and H. Kontani, Nematicity and magnetism in FeSe and other families of Fe-based super- conductors, Phys. Rev. X6, 021032 (2016)
work page 2016
- [21]
-
[22]
R. Fernandes, A. Chubukov, and J. Schmalian, What drives nematic order in iron-based superconductors, Nat. Phys.10, 97 (2014)
work page 2014
-
[23]
T. M. McQueen, A. J.Williams, P. Stephens, J. Tao, Y. Zhu, V. Ksenofontov, F. Casper, C. Felser, and R. J. Cava, Tetragonal-to-orthorhombic structural phase tran- sition at 90K in the superconductor Fe 1.01Se, Phys. Rev. Lett.103, 057002 (2009)
work page 2009
- [24]
-
[25]
M. Yi, D. H. Lu, R. Yu, S. C. Riggs, J.-H. Chu, B. Lv, Z. K. Liu, M. Lu, Y.-T. Cui, M. Hashimoto, S.-K. Mo, Z. Hussain, C. Chu, I. R. Fisher, Q. Si, and Z.-X. Shen, Observation of temperature-induced crossover to an orbital-selective mott phase in AFe2−ySe2 (A=K, Rb) superconductors, Phys. Rev. Lett.110, 067003 (2013)
work page 2013
-
[26]
Z. Yin, K. Haule, and G. Kotliar, Kinetic frustration and the nature of the magnetic and paramagnetic states in iron pnictides and iron chalcogenides, Nat. Mater.10, 932 (2011)
work page 2011
-
[27]
M. Ma, P. Bourges, Y. Sidis, Y. Xu, S. Li, B. Hu, J. Li, F. Wang, and Y. Li, Prominent role of spin-orbit coupling in FeSe revealed by inelastic neutron scattering, Phys. Rev. X7, 021025 (2017)
work page 2017
- [28]
- [29]
-
[30]
Z. Wang, X.-G. Zhao, R. Koch, S. J. L. Billinge, and A. Zunger, Understanding electronic peculiarities in tetragonal FeSe as local structural symmetry breaking, Phys. Rev. B102, 235121 (2020)
work page 2020
- [31]
-
[32]
J. M. Tomczak, M. van Schilfgaarde, and G. Kotliar, Many-body effects in iron pnictides and chalcogenides: Nonlocal versus dynamic origin of effective masses, Phys. Rev. Lett.109, 237010 (2012)
work page 2012
-
[33]
X. Long, S. Zhang, F. Wang, and Z. Liu, A first-principle perspective on electronic nematicity in FeSe, Npj Quan- tum Mater.5, 50 (2020)
work page 2020
-
[34]
Y. Ding, M. Zeng, Q. Zheng, J. Zhang, D. Xu, W. Chen, C. Wang, S. Chen, Y. Xie, Y. Ding, S. Zheng, J. Zhao, P. Gao, and L. Fu, Bidirectional and reversible tuning of the interlayer spacing of two-dimensional materials, Nat. Comm.12, 5886 (2021)
work page 2021
-
[35]
J. Kolorenˇ c, S. Hu, and L. Mitas, Wave functions for quantum monate carlo calculations in solids: Orbitals from density functional theory with hybrid exchange- correlation functionals, Phys. Rev. B82, 115108 (2010)
work page 2010
-
[36]
J. Kolorenˇ c and L. Mitas, Applications of quantum monte carlo methods in condensed systems, Reports on Progress in Physics74, 026502 (2011)
work page 2011
-
[37]
M. Dubecky, L. Mitas, and P. Jurecka, Noncovalent in- teractions by quantum monte carlo, Chemical Reviews 116, 5188 (2016)
work page 2016
-
[38]
J. Kolorenˇ c and L. Mitas, Quantum monte carlo calcu- lations of structural properties of FeO under pressure, Phys. Rev. Lett.101, 185502 (2008)
work page 2008
- [39]
-
[40]
B. Busemeyer, M. Dagrada, S. Sorella, M. Casula, , and L. K. Wagner, Competing collinear magnetic struc- tures in superconducting FeSe by first-principles quan- tum monte carlo calculations, Phys. Rev. B94, 035108 (2016)
work page 2016
-
[41]
C. J. Umrigar, J. Toulouse, C. Filippi, S. Sorella, and R. G. Hennig, Alleviation of the fermion-sign problem by optimization of many-body wave functions, Phys. Rev. Lett.98, 110201 (2007)
work page 2007
-
[42]
W. M. C. Foulkes, L. Mitas, R. J. Needs, and G. Ra- jagopal, Quantum monte carlo simulations of solids, Rev. Mod. Phys.73, 33 (2001)
work page 2001
-
[43]
F. Becca and S. Sorella,Quantum Monte Carlo ap- proches for correlated systems(Cambridge University, Cambridge, UK, 2017)
work page 2017
-
[44]
J. B. Anderson, Quantum chemistry by random walk., J. Chem. Phys.65, 4121 (1976)
work page 1976
- [45]
- [46]
- [47]
-
[48]
B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson, and T. L. Windus, A new basis set exchange: An open, up-to-date resource for the molecular sciences commu- nity, J. Chem. Inf. Model.59, 4814 (2019)
work page 2019
-
[49]
A. Annaberdiyev, G. Wang, C. A. Melton, M. C. Ben- nett, L. Shulenburger, and L. Mitas, A new generation of effective core potentials from correlated calculations: 3d transition metal series, J. Chem. Phys.149, 134108 (2018)
work page 2018
-
[50]
M. C. Bennett, C. A. Melton, A. Annaberdiyev, G. Wang, L. Shulenburger, and L. Mitas, A new genera- tion of effective core potentials for correlated calculations, J. Chem. Phys.147, 224106 (2017)
work page 2017
- [51]
-
[52]
J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B23, 5048 (1981)
work page 1981
-
[53]
S. Fahy, X. W. Wang, and S. G. Louie, Variational quantum monte carlo nonlocal pseudopotential approach to solids: Formulation and application to diamond, graphite, and silicon, Phys. Rev. B42, 3503 (1990)
work page 1990
-
[54]
D. M. Ceperley, Ground state of the fermion one- component plasma: A monte carlo study in two and three dimensions, Phys. Rev. B18, 3126 (1978)
work page 1978
-
[55]
Casula, Beyond the locality approximation in the standard diffusion monte carlo method, Phys
M. Casula, Beyond the locality approximation in the standard diffusion monte carlo method, Phys. Rev. B74, 161102(R) (2006)
work page 2006
- [56]
-
[57]
S. P. McCarthy and A. J. Thakkar, Accurate all-electron correlation energies for the closed-shell atoms from Ar to Rn and their relationship to the corresponding MP2 correlation energies, J. Chem. Phys.134, 044102 (2011)
work page 2011
-
[58]
J. R. Trail and R. J. Needs, Correlated electron pseu- dopotentials for 3d-transition metals, J. Chem. Phys. 142, 064110 (2015)
work page 2015
-
[59]
J. R. Trail and R. J. Needs, Shape and energy consis- tent pseudopotentials for correlated electron systems, J. Chem. Phys.146, 204107 (2017)
work page 2017
-
[60]
M. Burkatzki, C. Filippi, and M. Dolg, Energy-consistent pseudopotentials for qmc calculations, J. Chem. Phys. 126, 234105 (2007)
work page 2007
-
[61]
M. Burkatzki, C. Filippi, and M. Dolg, Energy-consistent small-core pseudopotentials for 3d-transition metals adapted to quantum monte carlo calculations, J. Chem. Phys.129, 164115 (2008)
work page 2008
-
[62]
”See Supplemental Material.”
-
[63]
S. Azadi and W. Foulkes, Systematic study of finite-size effects in quantum monte carlo calculations of real metal- lic systems, J. Chem. Phys.143, 102807 (2015)
work page 2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.