Recognition: unknown
Direct RNA sequence design under codon constraints using expressive tensor-based secondary structure models
Pith reviewed 2026-05-10 00:24 UTC · model grok-4.3
The pith
An algorithm directly samples codon sequences for a target protein from a Boltzmann distribution based on detailed RNA secondary structure energies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We demonstrate a direct and efficient algorithm to sample sequences from a suitable Boltzmann distribution defined in terms of the codon sequence and a fully detailed secondary structure free energy model, as well as related algorithms for exact computation of statistical quantities such as free energies, base pairing probabilities, and base and codon marginals. These draw upon a tensor-based formulation of secondary structure thermodynamics and show that global sequence design can be accomplished with respect to a highly accurate free energy model while leveraging CPU and GPU resources in parallel.
What carries the argument
The tensor-based formulation of secondary structure thermodynamics, extended to codon-constrained sequences to allow efficient sampling and exact marginal computations.
If this is right
- Sequence design can now use the full accuracy of secondary structure free energy models instead of simplified objectives.
- Exact values for free energies, base-pairing probabilities, and codon marginals become computable for realistic design problems.
- Parallel hardware can be used to achieve large speedups in the design process.
- Applications such as vaccine and therapeutic mRNA design gain a more principled way to optimize sequences.
Where Pith is reading between the lines
- The method could be combined with experimental feedback to iteratively improve sequence designs for cellular stability.
- It may extend to other constraints like translation speed or immune evasion in mRNA constructs.
- Scalability tests on longer proteins would reveal if the tensor approach remains practical for genome-scale applications.
Load-bearing premise
The tensor-based secondary structure model remains accurate and tractable when applied to the space of sequences that only use valid codons for each amino acid.
What would settle it
If the base-pairing probabilities computed by the algorithm for a test codon sequence deviate significantly from those measured by structure-probing experiments on the corresponding RNA.
Figures
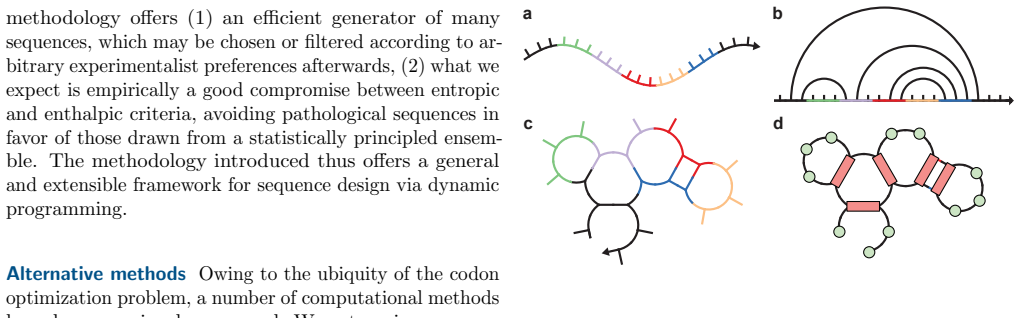
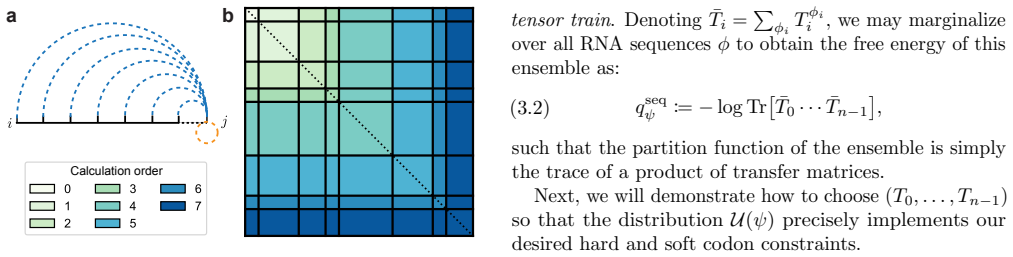




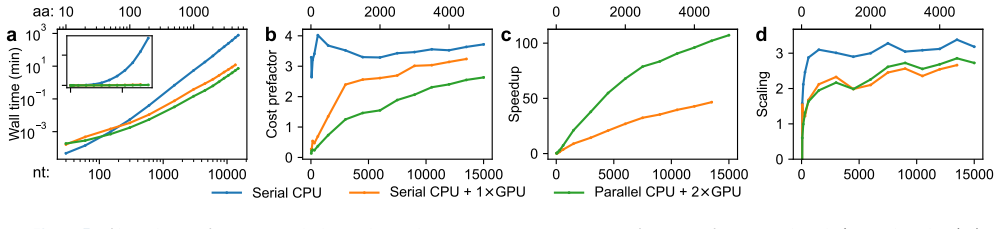











read the original abstract
Nucleic acid sequence design via codon optimization is a fundamental task with applications across synthetic biology, mRNA therapeutics, and vaccine design. Given a target protein, it is a major open challenge to navigate the combinatorially large design space of codon sequences mapping to its amino acid sequence. Computational approaches generally seek to optimize simple objectives based on the codon sequence, possibly together with more complicated contributions based on secondary structure analysis. In this work, we demonstrate a direct and efficient algorithm to sample sequences from a suitable Boltzmann distribution defined in terms of the codon sequence and a fully detailed secondary structure free energy model, as well as related algorithms for exact computation of statistical quantities such as free energies, base pairing probabilities, and base and codon marginals. These algorithms draw upon a recently developed tensor-based formulation of secondary structure thermodynamics and demonstrate, for the first time, that global sequence design can be accomplished with respect to a highly accurate free energy model. Moreover, the algorithms can leverage any available CPU and GPU resources in parallel for massive computational speedups.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents algorithms for direct RNA sequence design under codon constraints, using a tensor-based formulation of secondary structure thermodynamics. It claims to enable exact sampling of codon sequences from a Boltzmann distribution defined over a target amino acid sequence and a detailed free energy model, along with exact computation of partition functions, base-pairing probabilities, and base/codon marginals, with parallelization over CPU/GPU resources.
Significance. If the algorithmic extension is shown to preserve exactness and polynomial scaling, the work would advance codon optimization in synthetic biology and mRNA design by incorporating global secondary structure energetics directly, rather than relying on simplified or local objectives. The tensor approach and parallel scaling are noted strengths if substantiated.
major comments (2)
- [Abstract] Abstract: the claim that the tensor formulation extends to codon-constrained sequence spaces while remaining 'direct and efficient' and 'exact' is not supported by any recurrence, state-space analysis, or complexity bound. The per-position restriction to synonymous codons changes the effective alphabet and may require additional auxiliary states or tensor ranks; without explicit demonstration this remains a load-bearing gap for the central claim.
- No section provides small-instance verification, benchmark timings, or comparison against existing codon-optimization methods (e.g., those using simpler secondary-structure heuristics). The absence of any numerical results or implementation details prevents assessment of whether the claimed polynomial scaling and exact marginals are realized in practice.
minor comments (1)
- [Abstract] The abstract refers to 'expressive tensor-based secondary structure models' without clarifying what additional expressivity is gained over prior tensor formulations or how it interacts with the codon constraints.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive major comments. We address each point below and describe the revisions we will incorporate to address the concerns.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the tensor formulation extends to codon-constrained sequence spaces while remaining 'direct and efficient' and 'exact' is not supported by any recurrence, state-space analysis, or complexity bound. The per-position restriction to synonymous codons changes the effective alphabet and may require additional auxiliary states or tensor ranks; without explicit demonstration this remains a load-bearing gap for the central claim.
Authors: We agree that the abstract is too concise to convey the technical details of the extension. The full manuscript presents the tensor-based dynamic programming recursions in the Methods section, where the standard RNA folding recursions are adapted by restricting the nucleotide choices at each codon position to the synonymous codons corresponding to the fixed amino acid. Because the number of synonymous codons is bounded by a small constant (at most six), the state space and tensor ranks are unchanged from the unconstrained case, preserving both exactness and the O(n^3) asymptotic complexity. In the revision we will add an explicit subsection that states the modified recurrence relations, provides the state-space analysis, and derives the complexity bound, thereby substantiating the claims made in the abstract. revision: yes
-
Referee: [—] No section provides small-instance verification, benchmark timings, or comparison against existing codon-optimization methods (e.g., those using simpler secondary-structure heuristics). The absence of any numerical results or implementation details prevents assessment of whether the claimed polynomial scaling and exact marginals are realized in practice.
Authors: The present manuscript is primarily algorithmic and focuses on establishing exactness and polynomial scaling through the tensor formulation. We acknowledge that the lack of empirical results makes it difficult to evaluate practical performance. In the revised version we will add a Results section containing (i) verification on small instances by comparing partition functions and marginals against exhaustive enumeration, (ii) wall-clock timings on CPU and GPU for sequences of increasing length to illustrate parallel scaling, and (iii) a comparison against a baseline codon-optimization method that uses a simpler minimum-free-energy heuristic. Pseudocode and implementation notes will also be included in the supplement. revision: yes
Circularity Check
No circularity in claimed derivation; new algorithm extends prior tensor model independently
full rationale
The paper's central contribution is the demonstration of a direct algorithm for sampling codon sequences from a Boltzmann distribution over secondary structure free energies, together with exact marginal and probability computations. This is explicitly positioned as an extension of a recently developed tensor-based dynamic programming formulation to the codon-constrained alphabet. The tensor model is treated as an external prior (cited as 'recently developed'), and the new work consists of algorithmic modifications to enforce per-position codon restrictions while preserving exactness and polynomial scaling. No equation or claim reduces the output distribution, free energies, or marginals to a redefinition or statistical fit of the inputs; the derivation chain is the construction of the constrained DP recurrences themselves. The abstract and described claims contain no self-definitional loops, fitted-input predictions, or load-bearing self-citations that collapse the result to prior outputs by construction.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The recently developed tensor formulation correctly computes the partition function and derived quantities for secondary structure thermodynamics.
- domain assumption Boltzmann distribution over codon sequences and structures is the right objective for sequence design.
Forward citations
Cited by 1 Pith paper
-
GoForth: Language Models for RNA Design under Structure, Sequence, and Coding Constraints
GoForth is a forward-trained encoder-decoder RNA language model that generates sequences under mixed constraints on fold, sequence, and coding by separating sequence prior, forward folding sampler, and reward oracle.
Reference graph
Works this paper leans on
-
[1]
Alexaki, J
A. Alexaki, J. Kames et al. Codon and codon-pair usage tables (CoCoPUTs): Facilitating genetic varia- tion analyses and recombinant gene design.J. Mol. Biol., 431:2434–2441, 2019
2019
-
[2]
Athey, A
J. Athey, A. Alexaki et al. A new and updated resource for codon usage tables.BMC Bioinformatics, 18:391, 2017
2017
-
[3]
B. K.-S. Chung and D.-Y. Lee. Computational codon optimization of synthetic gene for protein expression. BMC Syst. Biol., 6:134, 2012
2012
-
[4]
B. Cohen and S. Skiena. Natural selection and algorithmic design of mRNA.Journal of Com- putational Biology, 10(3–4):419–432, 2003. doi: 10.1089/10665270360688101
-
[5]
J. R. Coleman, D. Papamichail et al. Virus atten- uation by genome-scale changes in codon pair bias. Science, 320:1784–1787, 2008
2008
-
[6]
Condon and C
A. Condon and C. Thachuk. Efficient codon op- timization with motif engineering.J. of Discrete Algorithms, 16:104–112, Oct. 2012. ISSN 1570-
2012
-
[7]
URL https: //doi.org/10.1016/j.jda.2012.04.017
doi: 10.1016/j.jda.2012.04.017. URL https: //doi.org/10.1016/j.jda.2012.04.017
-
[8]
D. A. Constant, J. M. Gutierrez et al. Deep learning- based codon optimization with large-scale synony- mous variant datasets enables generalized tunable protein expression.bioRxiv, 2023
2023
-
[9]
N. Dai, T. Zhou et al. EnsembleDesign: mes- senger RNA design minimizing ensemble free en- ergy via probabilistic lattice parsing.Bioinfor- matics, 41:i391–i400, 07 2025. ISSN 1367-4811. doi: 10.1093/bioinformatics/btaf245. URL https: //doi.org/10.1093/bioinformatics/btaf245
-
[10]
E. A. Demissie, S.-Y. Park et al. Comparative analysis of codon optimization tools: Advancing to- ward a multi-criteria framework for synthetic gene design.Journal of Microbiology and Biotechnol- ogy, 35:e2411066, Apr 2025. ISSN 1738-8872. doi: 10.4014/jmb.2411.11066. URL https://doi.org/10. 4014/jmb.2411.11066
-
[11]
Ding and C
Y. Ding and C. E. Lawrence. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res., 31:7280–7301, 2003
2003
-
[12]
R. M. Dirks, J. S. Bois et al. Thermodynamic analysis of interacting nucleic acid strands.SIAM Rev., 49: 65–88, 2007
2007
-
[13]
Elazar, S
A. Elazar, S. M. D A, and M. Madan Babu. Interro- gating nucleotide sequences with AI to understand codon usage patterns.Proc. Natl. Acad. Sci. U. S. A., 122:e2426326122, 2025
2025
-
[14]
M. Faizi, H. Sakharova, and L. F. Lareau. A genera- tive language model decodes contextual constraints on codon choice for mRNA design.bioRxiv, page 2025.05.13.653614, June 2025. doi: 10.1101/2025.05. 13.653614. URL https://doi.org/10.1101/2025.05.13. 653614. Preprint
-
[15]
Fallahpour, V
A. Fallahpour, V. Gureghian et al. Codontransformer: a multispecies codon optimizer using context-aware neural networks.Nature Communications, 16(1): 3205, Apr 2025. ISSN 2041-1723. doi: 10.1038/ s41467-025-58588-7. URL https://doi.org/10.1038/ s41467-025-58588-7
2025
-
[16]
Fornace and N
M. Fornace and N. A. Pierce. A new class of tensor- based models for nucleic acid secondary structure thermodynamics.in prep, 1, 2026
2026
-
[17]
M. E. Fornace, N. J. Porubsky, and N. A. Pierce. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: Enhanced models, scalability, and speed.ACS Synth. Biol., 9: 2665–2678, 2020. PMID: 32910644
2020
-
[18]
M. E. Fornace, J. Huang et al. NUPACK: Analysis and design of nucleic acid structures, devices, and systems.ChemRxiv, 2022
2022
-
[19]
X. Gu, Y. Qi, and M. El-Kebir. DERNA enables Pareto optimal RNA design.Journal of Compu- tational Biology, 31(3):179–196, mar 2024. ISSN 1557-8666. doi: 10.1089/cmb.2023.0283
-
[20]
Gustafsson, S
C. Gustafsson, S. Govindarajan, and J. Minshull. Codon bias and heterologous protein expression. Trends Biotechnol., 22:346–353, 2004
2004
-
[21]
G. A. Gutman and G. W. Hatfield. Nonrandom utilization of codon pairs in escherichia coli.Pro- ceedings of the National Academy of Sciences of the United States of America, 86(10):3699–3703, May 9
-
[22]
ISSN 0027-8424. doi: 10.1073/pnas.86.10.3699. URL https://research.ebsco.com/linkprocessor/plink? id=79060f14-94bd-3676-99a9-14d7f47cea52
-
[23]
N. K. Hegelmeyer, M. L. Previti et al. Gene recoding by synonymous mutations creates promiscuous intra- genic transcription initiation in mycobacteria.Mbio, 14, Mar. 2023
2023
-
[24]
L. Jin, Y. Zhou et al. mRNA vaccine sequence and structure design and optimization: Advances and challenges.Journal of Biological Chemistry, 301 (1), Jan 2025. ISSN 0021-9258. doi: 10.1016/j.jbc. 2024.108015. URL https://doi.org/10.1016/j.jbc.2024. 108015
-
[25]
pAN001-SY172 Wt- ATG, 2025
Joint BioEnergy Institute. pAN001-SY172 Wt- ATG, 2025. URL https://public-registry.jbei.org/ entry/21267. Accessed 15 Dec 2025 at https:// public-registry.jbei.org/entry/21267
2025
-
[26]
Kardar.Statistical physics of particles
M. Kardar.Statistical physics of particles. Cambridge University Press, 2007
2007
-
[27]
S. C. Kim, S. S. Sekhon et al. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency.Mol. Cell. Toxicol., 18:1–8, 2022
2022
-
[28]
Y.-A. Kim, K. Mousavi et al. Computational design of mRNA vaccines.Vaccine, 42:1831–1840, 2024
2024
-
[29]
A. Kloczkowski, T. Z. Sen, and R. L. Jerni- gan. The transfer matrix method for lattice proteins–an application with cooperative interac- tions.Polymer, 45(2):707–716, 2004. ISSN 0032-3861. doi: https://doi.org/10.1016/j.polymer.2003.10.072. URL https://www.sciencedirect.com/science/article/ pii/S0032386103010085. Conformational Protein Con- formations
-
[30]
M. S. D. Kormann, G. Hasenpusch et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice.Nat. Biotechnol., 29:154– 157, 2011
2011
-
[31]
M. J. Lajoie, A. J. Rovner et al. Genomically recoded organisms expand biological functions.Science, 342: 357–360, 2013
2013
-
[32]
R. A. Larocca, P. Abbink et al. Vaccine protection against Zika virus from Brazil.Nature, 536:474–478, 2016
2016
-
[33]
J. Li, H.-S. Lai et al. Arcade: Controllable codon design from foundation models via activation engi- neering.bioRxiv, Nov. 2025. doi: 10.1101/2025.08. 19.668819. URL https://doi.org/10.1101/2025.08.19. 668819. Preprint
-
[34]
Y. Li, F. Wang et al. Deep generative optimiza- tion of mRNA codon sequences for enhanced mRNA translation and therapeutic efficacy.Nature Com- munications, 16(1):9957, Nov 2025. ISSN 2041-
2025
-
[35]
URL https: //doi.org/10.1038/s41467-025-64894-x
doi: 10.1038/s41467-025-64894-x. URL https: //doi.org/10.1038/s41467-025-64894-x
-
[36]
J.-G. Liu, L. Wang, and P. Zhang. Tropical tensor network for ground states of spin glasses.Phys. Rev. Lett., 126:090506, 2021
2021
-
[37]
P. Mali, L. Yang et al. RNA-guided human genome engineering via Cas9.Science, 339:823–826, 2013
2013
-
[38]
Y. Mao, H. Liu et al. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in saccharomyces cerevisiae. Nucleic Acids Res., 42:4813–4822, 2014
2014
-
[39]
D. Mart´ ınez-Flores, J. Zepeda-Cervantes et al. SARS- CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants.Frontiers in Immunology, 12:701501, jul 2021. ISSN 1664-3224. doi: 10.3389/fimmu.2021.701501
-
[40]
D. H. Mathews, J. Sabina et al. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure.J. Mol. Biol., 288:911–940, 1999
1999
-
[41]
D. M. Mauger, B. J. Cabral et al. mRNA structure regulates protein expression through changes in func- tional half-life.Proc. Natl. Acad. Sci. U. S. A., 116: 24075–24083, 2019
2019
-
[42]
J. S. McCaskill. The equilibrium partition function and base pair binding probabilities for RNA sec- ondary structure.Biopolymers: Original Research on Biomolecules, 29:1105–1119, 1990
1990
-
[43]
R. A. Mir, J. Lovelace et al. Biophysical character- ization and modeling of human ecdysoneless (ecd) protein supports a scaffolding function.AIMS Bio- physics, 3(1):195–208, mar 2016. ISSN 2377-9098. doi: 10.3934/biophy.2016.1.195
-
[44]
I. V. Oseledets. Tensor-train decomposition.SIAM Journal on Scientific Computing, 33(5):2295–2317, 2011
2011
-
[45]
C. J. Paddon, P. J. Westfall et al. High-level semi-synthetic production of the potent antimalarial artemisinin.Nature, 496:528–532, 2013
2013
-
[46]
J.-E. Pin. Tropical semirings.Idempotency (Bristol, 1994), pages 50–69, 1998
1994
-
[47]
Z. Ren, L. Jiang et al. Codonbert: a bert-based architecture tailored for codon optimization us- ing the cross-attention mechanism.Bioinformat- ics, 40(7):btae330, 05 2024. ISSN 1367-4811. doi: 10.1093/bioinformatics/btae330. URL https://doi. org/10.1093/bioinformatics/btae330
-
[48]
Ringn´ er and M
M. Ringn´ er and M. Krogh. Folding free energies of 5’-UTRs impact post-transcriptional regulation on a genomic scale in yeast.PLoS Comput. Biol., 1:e72, 2005
2005
-
[49]
SantaLucia
J. SantaLucia. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermo- dynamics.Proceedings of the National Academy of Sciences, 95:1460–1465, 1998. 10
1998
-
[50]
Schmidt, N
M. Schmidt, N. Lee et al. Maximizing heterologous expression of engineered type I polyketide synthases: Investigating codon optimization strategies.ACS Synth. Biol., 12:3366–3380, 2023
2023
-
[51]
A. Sen, K. Kargar et al. Codon optimization: a mathematical programing approach.Bioinformatics, 36(13):4012–4020, 04 2020. ISSN 1367-4803. doi: 10.1093/bioinformatics/btaa248. URL https://doi. org/10.1093/bioinformatics/btaa248
-
[52]
M. J. Serra and D. H. Turner. Predicting thermody- namic properties of RNA.Methods Enzymol., 259: 242, 1995
1995
-
[53]
P. M. Sharp and W. H. Li. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications.Nucleic Acids Res, 15(3):1281–1295, Feb. 1987
1987
-
[54]
T. Sidi, S. Bahiri-Elitzur et al. Predicting gene sequences with AI to study codon usage patterns. Proceedings of the National Academy of Sciences of the United States of America, 122(1):e2410003121, jan
-
[55]
ISSN 1091-6490. doi: 10.1073/pnas.2410003121
-
[56]
Terai, S
G. Terai, S. Kamegai, and K. Asai. CDSfold: an algorithm for designing a protein-coding sequence with the most stable secondary structure.Bioin- formatics, 32(6):828–834, 11 2015. ISSN 1367-
2015
-
[57]
URL https://doi.org/10.1093/bioinformatics/btv678
doi: 10.1093/bioinformatics/btv678. URL https://doi.org/10.1093/bioinformatics/btv678
-
[58]
R. Thomas, G. Al-Khadairi et al. NY-ESO-1 based immunotherapy of cancer: Current perspectives.Fron- tiers in Immunology, 9:947, may 2018. ISSN 1664-3224. doi: 10.3389/fimmu.2018.00947
-
[59]
Tinoco, O
I. Tinoco, O. C. Uhlenbeck, and M. D. Levine. Esti- mation of secondary structure in ribonucleic acids. Nature, 230:362–367, 1971
1971
-
[60]
D. H. Turner and D. H. Mathews. NNDB: The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure.Nucleic Acids Res., 38:D280–D282, 2010
2010
-
[61]
O95905 ·ECD HUMAN, 2025
UniProt Consortium. O95905 ·ECD HUMAN, 2025. URL https://www.uniprot.org/uniprotkb/O95905/ entry. Accessed 15 Dec 2025 at https://www.uniprot. org/uniprotkb/O95905/entry
2025
-
[62]
P0DTC2 ·SPIKE SARS2, 2025
UniProt Consortium. P0DTC2 ·SPIKE SARS2, 2025. URL https://www.uniprot.org/uniprotkb/P0DTC2/ entry. Accessed 15 Dec 2025 at https://www.uniprot. org/uniprotkb/P0DTC2/entry
2025
-
[63]
P78358·CTG1B HUMAN, 2025
UniProt Consortium. P78358·CTG1B HUMAN, 2025. URL https://www.uniprot.org/uniprotkb/P78358/ entry. Accessed 15 Dec 2025 at https://www.uniprot. org/uniprotkb/P78358/entry
2025
-
[64]
A. B. Vogel, I. Kanevsky et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2.Nature, 592:283–289, 2021
2021
-
[65]
N. Vostrosablin, S. Lim et al. mRNAid, an open- source platform for therapeutic mRNA design and optimization strategies.NAR Genomics and Bioin- formatics, 6(1):lqae028, 03 2024. ISSN 2631-9268. doi: 10.1093/nargab/lqae028. URL https://doi.org/ 10.1093/nargab/lqae028
-
[66]
H. F. Walker and P. Ni. Anderson acceleration for fixed-point iterations.SIAM J. Numer. Anal., 49: 1715–1735, 2011
2011
-
[67]
M. Ward, M. Richardson, and M. Metkar. mRNA folding algorithms for structure and codon optimiza- tion.Briefings in Bioinformatics, 26(4):bbaf386, 08
-
[68]
ISSN 1477-4054. doi: 10.1093/bib/bbaf386. URL https://doi.org/10.1093/bib/bbaf386
-
[69]
M. Ward, M. Richardson, and M. Metkar. mRNA fold- ing algorithms for structure and codon optimization. arXiv [q-bio.BM], 2025
2025
-
[70]
H. K. Wayment-Steele, D. S. Kim et al. Theoret- ical basis for stabilizing messenger RNA through secondary structure design.Nucleic acids research, 49:10604–10617, 2021
2021
-
[71]
B. R. Wolfe, N. J. Porubsky et al. Constrained multistate sequence design for nucleic acid reaction pathway engineering.J. Am. Chem. Soc., 139:3134– 3144, 2017
2017
-
[72]
J. N. Zadeh, B. R. Wolfe, and N. A. Pierce. Nucleic acid sequence design via efficient ensemble defect optimization.J. Comput. Chem., 32:439–452, 2011
2011
-
[73]
Zhang, L
H. Zhang, L. Zhang et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature, 621:396–403, 2023
2023
-
[74]
Zhang, H
H. Zhang, H. Liu et al. Deep generative models design mRNA sequences with enhanced translational capacity and stability.Science, 390(6773):eadr8470,
-
[75]
URL https:// www.science.org/doi/abs/10.1126/science.adr8470
doi: 10.1126/science.adr8470. URL https:// www.science.org/doi/abs/10.1126/science.adr8470
-
[76]
Zuker and P
M. Zuker and P. Stiegler. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information.Nucleic Acids Res., 9:133–148, 1981
1981
-
[77]
Zulkower and S
V. Zulkower and S. Rosser. DNA chisel, a versatile sequence optimizer.Bioinformatics, 36:4508–4509, 2020. 11 Appendix S1. Glossary of symbols .We provide a glossary of notation below. Symbol Definition ψ the amino acid sequence (of length|ψ|) being coded for (a fixed input of our algorithms) ϕ an RNA sequence of lengthn(so thatn=|ϕ|), generally satisfying...
2020
-
[78]
For each unpaired basei, a matrix ˜Vi
-
[79]
For each base pairi·j, a pair of tensors ˜Bi,j and ˜Bj,i modeling each side of the base pair
-
[80]
A matrix Z modeling each strand break in secondary structure, constructed as Z = lr⊤ for global vectors l and r
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.