Recognition: no theorem link
Scalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins
Pith reviewed 2026-05-16 22:02 UTC · model grok-4.3
The pith
A tournament-based multi-agent system designs biologics for disordered proteins that outperform human references in over half of tested cases.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
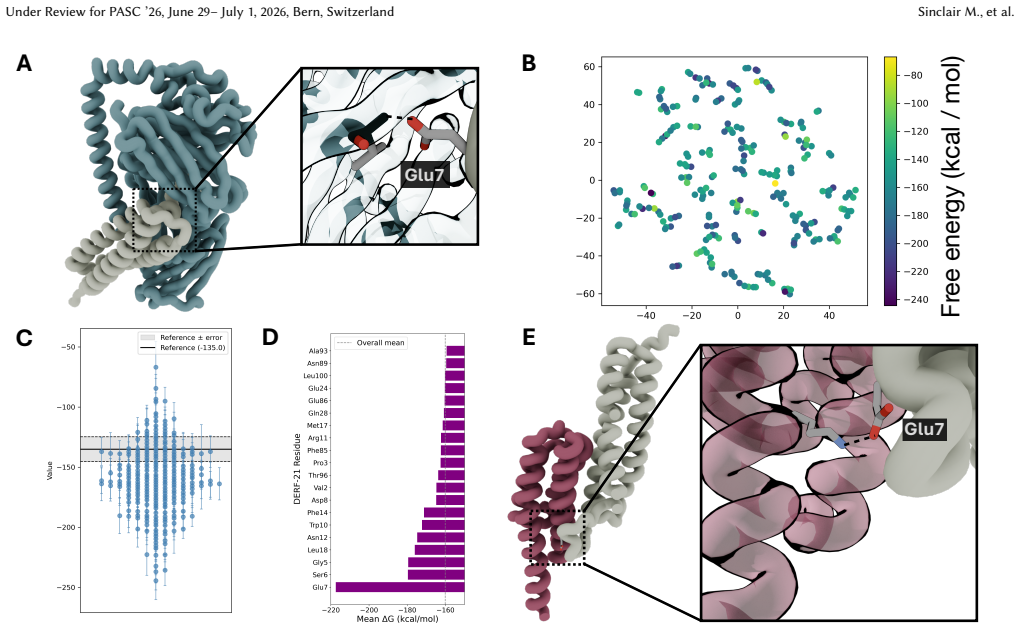
StructBioReasoner uses a novel tournament-based reasoning framework in which specialized agents compete to generate and refine therapeutic hypotheses for IDP binders. The agents coordinate literature synthesis, AI-structure prediction, molecular simulations, and stability analysis on HPC infrastructure via the Academy middleware. On Der f 21 more than 50 percent of 787 designed and validated candidates exceeded the binding free energy of human-designed reference binders from the literature. On NMNAT-2 the system identified three binding modes among 97,066 candidates, including the well-studied NMNAT2:p53 interface.
What carries the argument
The tournament-based reasoning framework in which specialized agents compete to generate and refine therapeutic hypotheses while orchestrating AI predictions, simulations, and stability analysis.
If this is right
- The system can explore design spaces containing tens of thousands of candidates for a single disordered protein target.
- Competitive agent interaction naturally distributes computational load across heterogeneous tools and hardware.
- Integration of multiple predictive modules produces multiple plausible binding modes rather than a single hypothesis.
- The same framework can be extended to larger Exascale platforms for broader therapeutic screening.
Where Pith is reading between the lines
- Similar tournament coordination might reduce human bias when designing binders for other flexible or multi-conformation targets.
- The recovered binding modes could be used as starting points for further experimental optimization or fragment-based elaboration.
- Scaling the agent count and tool diversity could enable simultaneous design against several IDP targets in one run.
Load-bearing premise
The computational predictions of binding free energy and binding modes accurately reflect real-world performance and the tournament process produces genuinely superior designs without post-hoc selection biases.
What would settle it
Laboratory binding-affinity measurements on a random sample of the designed candidates that show no statistical improvement over the human reference binders.
Figures




read the original abstract
Intrinsically disordered proteins (IDPs) represent crucial therapeutic targets due to their significant role in disease -- approximately 80\% of cancer-related proteins contain long disordered regions -- but their lack of stable secondary/tertiary structures makes them "undruggable". While recent computational advances, such as diffusion models, can design high-affinity IDP binders, translating these to practical drug discovery requires autonomous systems capable of reasoning across complex conformational ensembles and orchestrating diverse computational tools at scale.To address this challenge, we designed and implemented StructBioReasoner, a scalable multi-agent system for designing biologics that can be used to target IDPs. StructBioReasoner employs a novel tournament-based reasoning framework where specialized agents compete to generate and refine therapeutic hypotheses, naturally distributing computational load for efficient exploration of the vast design space. Agents integrate domain knowledge with access to literature synthesis, AI-structure prediction, molecular simulations, and stability analysis, coordinating their execution on HPC infrastructure via an extensible federated agentic middleware, Academy. We benchmark StructBioReasoner across Der f 21 and NMNAT-2 and demonstrate that over 50\% of 787 designed and validated candidates for Der f 21 outperformed the human-designed reference binders from literature, in terms of improved binding free energy. For the more challenging NMNAT-2 protein, we identified three binding modes from 97,066 binders, including the well-studied NMNAT2:p53 interface. Thus, StructBioReasoner lays the groundwork for agentic reasoning systems for IDP therapeutic discovery on Exascale platforms.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces StructBioReasoner, a scalable multi-agent system that uses a novel tournament-based reasoning framework to design biologics targeting intrinsically disordered proteins (IDPs). Agents integrate literature synthesis, AI-based structure prediction, molecular simulations, and stability analysis, coordinated via the Academy middleware on HPC resources. Benchmarks on Der f 21 report that over 50% of 787 designed and validated candidates outperformed human-designed literature references in binding free energy; on the more challenging NMNAT-2 target the system identified three binding modes (including the known NMNAT2:p53 interface) from 97,066 generated binders.
Significance. If the computational performance claims hold under rigorous validation, the work would represent a meaningful advance in autonomous, scalable agentic systems for IDP therapeutic design—an area of high unmet need given that ~80% of cancer-related proteins contain long disordered regions. The tournament framework for hypothesis competition and the federated middleware for tool orchestration are concrete contributions that could be adopted by other structural-biology pipelines.
major comments (3)
- [Abstract and Results] Abstract and Results: The headline claim that >50% of 787 Der f 21 candidates outperform literature references rests entirely on in silico binding free energies. No experimental Kd, SPR, ITC, or cell-based assay data are reported for any of the designs. For IDPs, where conformational ensembles render standard docking and MM-PBSA/GBSA estimates prone to large systematic errors, this absence is load-bearing for the central performance assertion.
- [Methods] Methods: The manuscript provides no details on validation protocols, error estimation, candidate selection criteria, or the number of designs filtered before arriving at the final 787 candidates. Without these, it is impossible to determine whether the reported 50% success rate reflects genuine affinity gains or post-hoc selection biases in the tournament or downstream simulation steps.
- [Results (NMNAT-2)] Results (NMNAT-2 section): The identification of three binding modes from 97,066 binders, including the NMNAT2:p53 interface, lacks quantitative metrics on how the tournament framework ranks or filters modes and whether orthogonal simulation protocols (e.g., enhanced sampling) were used to mitigate IDP-specific artifacts.
minor comments (2)
- [Abstract] Abstract: The phrase 'designed and validated candidates' is used without a clear definition of what 'validated' means in the computational pipeline; add a concise clarification.
- [Figures and Methods] Figure captions and Methods: Ensure all tool versions, force-field parameters, and simulation lengths are explicitly stated so that the binding-free-energy calculations can be reproduced.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and positive assessment of the work's potential significance. We address each major comment point by point below, with revisions planned to improve clarity and rigor where appropriate.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results: The headline claim that >50% of 787 Der f 21 candidates outperform literature references rests entirely on in silico binding free energies. No experimental Kd, SPR, ITC, or cell-based assay data are reported for any of the designs. For IDPs, where conformational ensembles render standard docking and MM-PBSA/GBSA estimates prone to large systematic errors, this absence is load-bearing for the central performance assertion.
Authors: We agree that the reported performance is based solely on computational binding free energies and that experimental data would provide stronger validation. This manuscript focuses on the agentic system and its in silico benchmarking using established protocols (multi-replica MM-PBSA/GBSA with ensemble averaging). We will revise the abstract and results to explicitly frame all metrics as computational estimates, add a dedicated limitations paragraph discussing IDP-specific challenges with these methods, and outline future experimental plans. This provides necessary context without overstating current claims. revision: partial
-
Referee: [Methods] Methods: The manuscript provides no details on validation protocols, error estimation, candidate selection criteria, or the number of designs filtered before arriving at the final 787 candidates. Without these, it is impossible to determine whether the reported 50% success rate reflects genuine affinity gains or post-hoc selection biases in the tournament or downstream simulation steps.
Authors: We will substantially expand the Methods section to include: full validation protocols (10-replica simulations per complex), error estimation (standard deviation across replicas plus bootstrapping), explicit candidate selection and filtering criteria (binding energy thresholds, stability scores, and tournament advancement rules), and the complete pipeline showing reduction from initial generations to the final 787 validated candidates. These additions will allow readers to evaluate potential biases directly. revision: yes
-
Referee: [Results (NMNAT-2)] Results (NMNAT-2 section): The identification of three binding modes from 97,066 binders, including the NMNAT2:p53 interface, lacks quantitative metrics on how the tournament framework ranks or filters modes and whether orthogonal simulation protocols (e.g., enhanced sampling) were used to mitigate IDP-specific artifacts.
Authors: We will update the NMNAT-2 results to report quantitative tournament metrics, including the scoring function, ranking thresholds, and fraction of candidates advanced at each stage. We will also specify that replica-exchange molecular dynamics (REMD) was employed as an orthogonal enhanced sampling method alongside standard simulations to address IDP conformational sampling, with consistency checks across protocols supporting the identified modes. revision: yes
- Absence of experimental binding data (Kd, SPR, etc.) for any designed candidates, which cannot be added without performing new wet-lab experiments outside the scope of this computational study.
Circularity Check
No significant circularity: performance claims derive from external simulation protocols applied to agent-generated designs, without self-referential fitting or definitional reduction.
full rationale
The paper presents StructBioReasoner as a multi-agent tournament system that generates candidate binders for IDPs, then evaluates them via AI structure prediction, molecular simulations, and binding free energy calculations. The central claim (>50% of 787 Der f 21 candidates outperforming literature references) is a direct numerical comparison of simulation outputs against external human-designed references. No equations, fitted parameters, or self-definitional loops are described; the binding free energy metric is computed by standard external tools (MM-PBSA/GBSA-style) whose implementation is not redefined by the agents. No self-citation chain justifies a uniqueness theorem or ansatz that forces the result. The tournament distributes exploration but does not statistically force the reported outperformance by construction, as the same simulation protocol is applied uniformly to both agent designs and literature references. This is an empirical systems paper whose validation chain remains independent of its own inputs.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption AI-based structure prediction tools provide reliable conformational ensembles for IDPs
- domain assumption Molecular simulations yield binding free energies that correlate with experimental outcomes
invented entities (2)
-
StructBioReasoner
no independent evidence
-
tournament-based reasoning framework
no independent evidence
Forward citations
Cited by 2 Pith papers
-
VibeProteinBench: An Evaluation Benchmark for Language-interfaced Vibe Protein Design
VibeProteinBench is a three-stage language-interfaced benchmark revealing that no current LLM performs strongly across recognition, engineering, and generation of proteins.
-
VibeProteinBench: An Evaluation Benchmark for Language-interfaced Vibe Protein Design
VibeProteinBench is a new benchmark evaluating LLMs on open-ended language-interfaced protein design across recognition, engineering, and generation, with no model showing strong performance in all areas.
Reference graph
Works this paper leans on
-
[1]
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. 2024. Accurate structure prediction of biomolecular interactions with AlphaFold 3.Nature630, 8016 (2024), 493–500
work page 2024
-
[2]
Sandhini Agarwal, Lama Ahmad, Jason Ai, Sam Altman, Andy Applebaum, Edwin Arbus, Rahul K Arora, Yu Bai, Bowen Baker, Haiming Bao, et al. 2025. gpt-oss-120b & gpt-oss-20b model card.arXiv preprint arXiv:2508.10925(2025)
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[3]
Gustaf Ahdritz, Nazim Bouatta, Christina Floristean, Sachin Kadyan, Qinghui Xia, William Gerecke, Timothy J O’Donnell, Daniel Berenberg, Ian Fisk, Niccolò Zanichelli, et al. 2024. OpenFold: Retraining AlphaFold2 yields new insights into Scalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins Under Review for PASC ’26...
work page 2024
-
[4]
Bryce Allen, John Bresnahan, Lisa Childers, Ian Foster, Gopi Kandaswamy, Raj Kettimuthu, Jack Kordas, Mike Link, Stuart Martin, Karl Pickett, et al . 2012. Software as a service for data scientists.Commun. ACM55, 2 (2012), 81–88
work page 2012
-
[5]
Benjamin S Allen, James Anchell, Victor Anisimov, Thomas Applencourt, Ab- hishek Bagusetty, Ramesh Balakrishnan, Riccardo Balin, Solomon Bekele, Colleen Bertoni, Cyrus Blackworth, et al . 2025. Aurora: Architecting Argonne’s First Exascale Supercomputer for Accelerated Scientific Discovery.arXiv preprint arXiv:2509.08207(2025)
-
[6]
Yadu Babuji, Anna Woodard, Zhuozhao Li, Daniel S Katz, Ben Clifford, Rohan Kumar, Lukasz Lacinski, Ryan Chard, Justin M Wozniak, Ian Foster, et al. 2019. Parsl: Pervasive parallel programming in Python. In28th International Symposium on High-Performance Parallel and Distributed Computing. 25–36
work page 2019
-
[7]
Daniil A Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes. 2023. Au- tonomous chemical research with large language models.Nature624, 7992 (2023), 570–578
work page 2023
-
[8]
Jacques Boitreaud, Jack Dent, Matthew McPartlon, Joshua Meier, Vinicius Reis, Alex Rogozhnikov, and Kevin Wu. 2024. Chai-1: Decoding the molecular interac- tions of life.BioRxiv(2024). doi:10.1101/2024.10.10.615955
-
[9]
Kaushik Borthakur, Thomas R Sisk, Francesco P Panei, Massimiliano Bonomi, and Paul Robustelli. 2025. Determining accurate conformational ensembles of intrinsically disordered proteins at atomic resolution.Nature Communications 16, 1 (2025), 9036
work page 2025
-
[10]
David A Case, Hasan Metin Aktulga, Kellon Belfon, David S Cerutti, G Andrés Cisneros, Vinícius Wilian D Cruzeiro, Negin Forouzesh, Timothy J Giese, An- dreas W Gotz, Holger Gohlke, et al . 2023. AmberTools.Journal of chemical information and modeling63, 20 (2023), 6183–6191
work page 2023
-
[11]
Bo Chen, Xingyi Cheng, Pan Li, Yangli-ao Geng, Jing Gong, Shen Li, Zhilei Bei, Xu Tan, Boyan Wang, Xin Zeng, et al . 2025. xTrimoPGLM: Unified 100- billion-parameter pretrained transformer for deciphering the language of proteins. Nature Methods(2025), 1–12
work page 2025
-
[12]
Anthony Costa. 2024. Better Molecules, Faster: NVIDIA Blueprint Re- defines Hit Identification With Generative AI-Based Virtual Screening — blogs.nvidia.com. https://blogs.nvidia.com/blog/nvidia-nim-agent-blueprint- virtual-screening/. Accessed 2025
work page 2024
-
[13]
2024.CrewAI: Role-Based Multi-Agent Framework for LLMs
CrewAI Developers. 2024.CrewAI: Role-Based Multi-Agent Framework for LLMs. https://github.com/joaomdmoura/crewAI Accessed 2025
work page 2024
-
[14]
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courbet, Rob J de Haas, Neville Bethel, et al. 2022. Robust deep learning–based protein sequence design using Protein- MPNN.Science378, 6615 (2022), 49–56
work page 2022
-
[15]
Kerry Dhakal. 2019. Unpaywall.Journal of the Medical Library Association107, 2 (2019), 286
work page 2019
-
[16]
Gautham Dharuman, Kyle Hippe, Alexander Brace, Sam Foreman, Väinö Hatan- pää, Varuni K Sastry, Huihuo Zheng, Logan Ward, Servesh Muralidharan, Archit Vasan, et al. 2024. MProt-DPO: Breaking the ExaFLOPS barrier for multimodal protein design workflows with direct preference optimization. InSC24: Inter- national Conference for High Performance Computing, Ne...
work page 2024
-
[17]
Gautham Dharuman, Logan Ward, Heng Ma, Priyanka V. Setty, Ozan Gokdemir, Sam Foreman, Murali Emani, Kyle Hippe, Alexander Brace, Kristopher Keipert, Thomas Gibbs, Ian Foster, Anima Anandkumar, Venkatram Vishwanath, and Arvind Ramanathan. 2023. Protein Generation via Genome-scale Language Models with Bio-physical Scoring. InProceedings of the SC ’23 Worksh...
-
[18]
Peter Eastman, Raimondas Galvelis, Raúl P Peláez, Charlles RA Abreu, Stephen E Farr, Emilio Gallicchio, Anton Gorenko, Michael M Henry, Frank Hu, Jing Huang, et al. 2023. OpenMM 8: Molecular dynamics simulation with machine learning potentials.The Journal of Physical Chemistry B128, 1 (2023), 109–116
work page 2023
-
[19]
Ian Foster. 2011. Globus Online: Accelerating and democratizing science through cloud-based services.IEEE Internet Computing15, 3 (2011), 70–73
work page 2011
-
[20]
Alireza Ghafarollahi and Markus J Buehler. 2024. ProtAgents: Protein discovery via large language model multi-agent collaborations combining physics and machine learning.Digital Discovery3, 7 (2024), 1389–1409
work page 2024
-
[21]
Ozan Gokdemir, Carlo Siebenschuh, Alexander Brace, Azton Wells, Brian Hsu, Kyle Hippe, Priyanka Setty, Aswathy Ajith, J Gregory Pauloski, Varuni Sastry, et al. 2025. HiPerRAG: High-Performance Retrieval Augmented Generation for Scientific Insights. InProceedings of the Platform for Advanced Scientific Computing Conference. 1–13
work page 2025
- [22]
-
[23]
2019.MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations
Richard J Gowers, Max Linke, Jonathan Barnoud, Tyler John Edward Reddy, Manuel N Melo, Sean L Seyler, Jan Domanski, David L Dotson, Sébastien Buchoux, Ian M Kenney, et al. 2019.MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. Technical Report. Los Alamos National Laboratory (LANL), Los Alamos, NM (United States)
work page 2019
-
[24]
Yu Gu, Robert Tinn, Hao Cheng, Michael Lucas, Naoto Usuyama, Xiaodong Liu, Tristan Naumann, Jianfeng Gao, and Hoifung Poon. 2021. Domain-specific language model pretraining for biomedical natural language processing.ACM Transactions on Computing for Healthcare (HEALTH)3, 1 (2021), 1–23
work page 2021
-
[25]
Marti A Hearst. 1997. Text tiling: Segmenting text into multi-paragraph subtopic passages.Computational Linguistics23, 1 (1997), 33–64
work page 1997
-
[26]
Ginny Hendricks, Dominika Tkaczyk, Jennifer Lin, and Patricia Feeney. 2020. Crossref: The sustainable source of community-owned scholarly metadata.Quan- titative Science Studies1, 1 (2020), 414–427
work page 2020
-
[27]
Alex S Holehouse and Birthe B Kragelund. 2024. The molecular basis for cellular function of intrinsically disordered protein regions.Nature Reviews Molecular Cell Biology25, 3 (2024), 187–211
work page 2024
-
[28]
Kexin Huang, Serena Zhang, Hanchen Wang, Yuanhao Qu, Yingzhou Lu, Yusuf Roohani, Ryan Li, Lin Qiu, Gavin Li, Junze Zhang, et al. 2025. Biomni: A general- purpose biomedical AI agent.biorxiv(2025). doi:10.1101/2025.05.30.656746
- [29]
-
[30]
IDEAL. 2024. AI-Driven Cancer Treatment and Vaccine Development. https: //www.anl.gov/cels/aidriven-cancer-treatment-and-vaccine-development. Ac- cessed 2025
work page 2024
-
[31]
John B Ingraham, Max Baranov, Zak Costello, Karl W Barber, Wujie Wang, Ahmed Ismail, Vincent Frappier, Dana M Lord, Christopher Ng-Thow-Hing, Erik R Van Vlack, et al. 2023. Illuminating protein space with a programmable generative model.Nature623, 7989 (2023), 1070–1078
work page 2023
-
[32]
Peter St John, Dejun Lin, Polina Binder, Malcolm Greaves, Vega Shah, John St John, Adrian Lange, Patrick Hsu, Rajesh Illango, Arvind Ramanathan, et al
- [33]
-
[34]
Jeff Johnson, Matthijs Douze, and Hervé Jégou. 2019. Billion-scale similarity search with GPUs.IEEE Transactions on Big Data7, 3 (2019), 535–547
work page 2019
-
[35]
2023.LangChain: Building Applications with Large Lan- guage Models
LangChain Developers. 2023.LangChain: Building Applications with Large Lan- guage Models. https://github.com/langchain-ai/langchain Accessed 2025
work page 2023
-
[36]
2024.LangGraph: Stateful, Multi-Agent LLM Workflows
LangChain Developers. 2024.LangGraph: Stateful, Multi-Agent LLM Workflows. https://github.com/langchain-ai/langgraph Accessed 2025
work page 2024
-
[37]
Patrick Lewis, Ethan Perez, Aleksandra Piktus, Fabio Petroni, Vladimir Karpukhin, Naman Goyal, Heinrich Küttler, Mike Lewis, Wen-tau Yih, Tim Rocktäschel, Sebastian Riedel, and Douwe Kiela. 2020. Retrieval-augmented generation for knowledge-intensive nlp tasks.Advances in neural information processing systems 33 (2020), 9459–9474
work page 2020
-
[38]
Caixuan Liu, Kejia Wu, Hojun Choi, Hannah L Han, Xueli Zhang, Joseph L Watson, Green Ahn, Jason Z Zhang, Sara Shijo, Lydia L Good, et al. 2025. Diffusing protein binders to intrinsically disordered proteins.Nature644, 8077 (2025), 809–817
work page 2025
- [39]
-
[40]
Jeffrey M. Lotthammer, Garrett M. Ginell, Daniel Griffith, Ryan J. Emenecker, and Alex S. Holehouse. 2024. Direct prediction of intrinsically disordered protein conformational properties from sequence.Nature Methods21, 3 (01 Mar 2024), 465–476
work page 2024
-
[41]
Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D White, and Philippe Schwaller
Andres M. Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D White, and Philippe Schwaller. 2024. Augmenting large language models with chemistry tools.Nature Machine Intelligence6, 5 (2024), 525–535
work page 2024
-
[42]
Robert T McGibbon, Kyle A Beauchamp, Matthew P Harrigan, Christoph Klein, Jason M Swails, Carlos X Hernández, Christian R Schwantes, Lee-Ping Wang, Thomas J Lane, and Vijay S Pande. 2015. MDTraj: A modern open library for the analysis of molecular dynamics trajectories.Biophysical journal109, 8 (2015), 1528–1532
work page 2015
-
[43]
Rui Meng, Ye Liu, Shafiq Rayhan Joty, Caiming Xiong, Yingbo Zhou, and Semih Yavuz. 2024. SFR-Embedding-Mistral:Enhance Text Retrieval with Transfer Learning. Salesforce AI Research Blog. https://www.salesforce.com/blog/sfr- embedding/
work page 2024
-
[44]
Bill R Miller III, T Dwight McGee Jr, Jason M Swails, Nadine Homeyer, Holger Gohlke, and Adrian E Roitberg. 2012. MMPBSA. py: an efficient program for end-state free energy calculations.Journal of chemical theory and computation8, 9 (2012), 3314–3321
work page 2012
- [45]
-
[46]
Alireza Omidi, Mads Harder Møller, Nawar Malhis, Jennifer M Bui, and Jörg Gsponer. 2024. AlphaFold-Multimer accurately captures interactions and dy- namics of intrinsically disordered protein regions.Proceedings of the National Academy of Sciences121, 44 (2024), e2406407121. Under Review for PASC ’26, June 29– July 1, 2026, Bern, Switzerland Sinclair M., et al
work page 2024
-
[47]
Martin Pacesa, Lennart Nickel, Christian Schellhaas, Joseph Schmidt, Ekaterina Pyatova, Lucas Kissling, Patrick Barendse, Jagrity Choudhury, Srajan Kapoor, Ana Alcaraz-Serna, et al. 2025. One-shot design of functional protein binders with BindCraft.Nature(2025), 1–10
work page 2025
-
[48]
Sze Lei Pang, Kok Lian Ho, Jitka Waterman, Robert Paul Rambo, Aik-Hong Teh, Indran Mathavan, Gemma Harris, Konstantinos Beis, Yee-How Say, Matta Sri Anusha, Yang Yie Sio, Fook tim Chew, and Chyan Leong Ng. 2019. Crystal structure and epitope analysis of house dust mite allergen Der f 21.Scientific Reports9, 1 (2019), 4933
work page 2019
-
[49]
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, et al
-
[50]
Boltz-2: Towards accurate and efficient binding affinity prediction.BioRxiv (2025). doi:10.1101/2025.06.14.659707
- [51]
-
[52]
Jason Priem, Heather Piwowar, and Richard Orr. 2022. OpenAlex: A fully-open index of scholarly works, authors, venues, institutions, and concepts.arXiv preprint arXiv:2205.01833(2022)
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[53]
Rafael Rafailov, Archit Sharma, Eric Mitchell, Christopher D Manning, Stefano Ermon, and Chelsea Finn. 2023. Direct preference optimization: Your language model is secretly a reward model.Advances in neural information processing systems36 (2023), 53728–53741
work page 2023
-
[54]
Jacob T Rapp, Bennett J Bremer, and Philip A Romero. 2024. Self-driving labora- tories to autonomously navigate the protein fitness landscape.Nature chemical engineering1, 1 (2024), 97–107
work page 2024
-
[55]
Summer Rosonovski, Maria Levchenko, Rajat Bhatnagar, Umamageswari Chan- drasekaran, Lynne Faulk, Islam Hassan, Matt Jeffryes, Syed Irtaza Mubashar, Maaly Nassar, Madhumiethaa Jayaprabha Palanisamy, Michael Parkin, Ja- gadeeswararao Poluru, Frances Rogers, Shyamasree Saha, Mohamed Selim, Zu- naira Shafique, Michele Ide-Smith, David Stephenson, Santosh Tiru...
work page 2024
- [56]
-
[57]
2025.molecular-simulations: A Python toolkit for MD simulation and analysis
Matt Sinclair and Moeen Meigooni. 2025.molecular-simulations: A Python toolkit for MD simulation and analysis. https://github.com/msinclair-py/molecular- simulations
work page 2025
-
[58]
Kyle Swanson, Wesley Wu, Nash L Bulaong, John E Pak, and James Zou. 2025. The Virtual Lab of AI agents designs new SARS-CoV-2 nanobodies.Nature646, 8085 (2025), 716–723
work page 2025
-
[59]
Colin S Swenson, Gunasheil Mandava, Deborah M Thomas, and Raymond E Moellering. 2024. Tackling undruggable targets with designer peptidomimetics and synthetic biologics.Chemical Reviews124, 22 (2024), 13020–13093
work page 2024
-
[60]
Chuan Tian, Koushik Kasavajhala, Kellon AA Belfon, Lauren Raguette, He Huang, Angela N Migues, John Bickel, Yuzhang Wang, Jorge Pincay, Qin Wu, et al
-
[61]
ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution.Journal of chemical theory and computation16, 1 (2019), 528–552
work page 2019
-
[62]
Masao Utiyama and Hitoshi Isahara. 2001. A statistical model for domain- independent text segmentation. In39th Annual Meeting of the Association for Computational Linguistics. 499–506
work page 2001
-
[63]
Twan Van Laarhoven. 2017. L2 regularization versus batch and weight normal- ization.arXiv preprint arXiv:1706.05350(2017)
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[64]
Archit Vasan, Ozan Gokdemir, Alexander Brace, Arvind Ramanathan, Thomas Brettin, Rick Stevens, and Venkatram Vishwanath. 2024. High Performance Bind- ing Affinity Prediction with a Transformer-Based Surrogate Model. In2024 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW). 571–580. doi:10.1109/IPDPSW63119.2024.00114
-
[65]
Hanping Wang, Ruoyao Xiong, and Luhua Lai. 2023. Rational drug design targeting intrinsically disordered proteins.Wiley Interdisciplinary Reviews: Com- putational Molecular Science13, 6 (2023), e1685
work page 2023
-
[66]
Yuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel Bautista
- [67]
-
[68]
Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borst, Robert J Ragotte, Lukas F Milles, et al. 2023. De novo design of protein structure and function with RFdiffusion. Nature620, 7976 (2023), 1089–1100
work page 2023
-
[69]
Kejia Wu, Hanlun Jiang, Derrick R Hicks, Caixuan Liu, Edin Muratspahić, Theresa A Ramelot, Yuexuan Liu, Kerrie McNally, Sebastian Kenny, Andrei Mihut, et al. 2025. Design of intrinsically disordered region binding proteins.Science 389, 6757 (2025), eadr8063
work page 2025
-
[70]
Junjie Zhu, Zhengxin Li, Zhuoqi Zheng, Bo Zhang, Bozitao Zhong, Jie Bai, Xi- aokun Hong, Taifeng Wang, Ting Wei, Jianyi Yang, et al. 2025. Accurate Gener- ation of Conformational Ensembles for Intrinsically Disordered Proteins with IDPFold.Advanced Science(2025), e11636
work page 2025
-
[71]
Maxim Zvyagin, Alexander Brace, Kyle Hippe, Yuntian Deng, Bin Zhang, Cindy Orozco Bohorquez, Austin Clyde, Bharat Kale, Danilo Perez-Rivera, Heng Ma, et al. 2023. GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics.The International Journal of High Performance Computing Applications37, 6 (2023), 683–705. Moeen Meigooni1„ Archit V...
work page 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.