CRANE: Correcting Errors in Raw Nanopore Signals Using Hidden Markov Models
Pith reviewed 2026-05-21 11:02 UTC · model grok-4.3
The pith
A Hidden Markov Model corrects errors in raw nanopore signals to raise the accuracy of direct signal analysis tools.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
CRANE trains and utilizes a Hidden Markov Model to accurately correct signal errors in raw nanopore data, which consistently improves the overall accuracy of raw signal analysis tools, minimizes the burden of optimizing analysis pipelines for newer nanopore technologies, and does not introduce substantial computational overhead.
What carries the argument
Hidden Markov Model trained on raw current signals to identify and correct error-prone transitions before downstream analysis.
If this is right
- Raw-signal mapping and alignment tools produce higher-accuracy results after the correction step.
- Analysis pipelines need less manual retuning when new nanopore pore versions or chemistries are introduced.
- The added runtime cost remains small relative to the accuracy improvement.
- The method supports development of error-correction techniques designed specifically for raw signals rather than base-called sequences.
Where Pith is reading between the lines
- The same HMM correction idea could be tested on raw signals from other long-read technologies that produce noisy current or optical traces.
- Real-time deployment during a sequencing run might allow error correction to occur as the molecule translocates.
- A small amount of new data from an unseen pore could be used to fine-tune the existing HMM and check whether generalization improves further.
Load-bearing premise
The dominant error modes in raw nanopore current signals are sufficiently stationary and Markovian that a single HMM trained on existing datasets will generalize to new molecules, new pore chemistries, and new analysis tools without retraining.
What would settle it
Applying the trained HMM to raw signals from a new pore chemistry or a different analysis tool and observing no accuracy gain or a loss would falsify the generalization claim.
Figures

read the original abstract
Nanopore sequencing can read substantially longer sequences of nucleic acid molecules, called reads, than other sequencing methods, which has led to advances in genomic analysis such as the gapless human genome assembly. By analyzing the raw electrical signal reads that nanopore sequencing generates from molecules, existing works can map these reads without translating them into DNA characters (i.e., basecalling), allowing for quick and efficient analysis of sequencing data. However, raw signals often contain errors due to noise and processing errors, which limits the overall accuracy of raw signal analysis. Our goal in this work is to detect and correct errors in raw signals to improve the accuracy of raw signal analyses. To this end, we propose CRANE, a mechanism that trains and utilizes a Hidden Markov Model (HMM) to accurately correct signal errors. Our extensive evaluation on various datasets shows that CRANE 1) consistently improves the overall accuracy of the underlying raw signal analysis tools, 2) minimizes the burden of optimizing analysis pipelines for newer nanopore technologies, and 3) does not introduce substantial computational overhead. We conclude that CRANE provides an effective mechanism to systematically identify and correct the errors in raw nanopore signals before further analysis, which can enable the development of a new class of error correction mechanisms purely designed for raw nanopore signals. Source Code: CRANE is available at https://github.com/STORMgroup/CRANE. We also provide the scripts to fully reproduce our results on our GitHub page
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes CRANE, a Hidden Markov Model (HMM) trained on existing nanopore datasets to detect and correct errors in raw electrical current signals prior to downstream analysis. The central claims are that this correction consistently improves accuracy of raw-signal tools, reduces the need to retune analysis pipelines when new pore chemistries or molecules appear, and adds negligible computational cost; the method is evaluated on held-out datasets and source code is provided for reproducibility.
Significance. If the quantitative claims are substantiated, CRANE would supply a practical, basecaller-agnostic preprocessing step that could lower the engineering burden of adapting raw-signal pipelines to successive nanopore chemistries. The reproducibility artifacts (public code and scripts) are a clear strength.
major comments (2)
- [Abstract / Evaluation] Abstract and Evaluation sections: the headline assertions of 'consistent improvements,' 'minimizes the burden,' and 'does not introduce substantial computational overhead' are presented without any reported accuracy deltas, error bars, dataset sizes, number of replicates, or ablation controls. Because these metrics are load-bearing for all three claims, their absence prevents assessment of statistical robustness or practical effect size.
- [Method / Evaluation] Method and Evaluation sections: the core modeling assumption—that a single HMM whose transition and emission parameters are fitted once on existing data will remain effective for new molecules, new pore chemistries, and new downstream tools without retraining or architectural change—is not accompanied by explicit cross-chemistry or cross-tool transfer experiments. Nanopore current statistics are known to shift with pore chemistry; if those shifts dominate, the correction step could degrade rather than improve accuracy, directly undermining the 'minimizes burden' claim.
minor comments (2)
- [Abstract] Abstract: consider adding one or two concrete performance numbers (e.g., 'X % relative improvement on dataset Y') to give readers an immediate sense of scale.
- [Methods] The manuscript would benefit from a short table summarizing the HMM state space, number of free parameters, and training-set size.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed feedback on the manuscript. We address each major comment below and have revised the manuscript to improve the substantiation and clarity of the reported claims.
read point-by-point responses
-
Referee: [Abstract / Evaluation] Abstract and Evaluation sections: the headline assertions of 'consistent improvements,' 'minimizes the burden,' and 'does not introduce substantial computational overhead' are presented without any reported accuracy deltas, error bars, dataset sizes, number of replicates, or ablation controls. Because these metrics are load-bearing for all three claims, their absence prevents assessment of statistical robustness or practical effect size.
Authors: We agree that the absence of specific quantitative metrics in the abstract and a consolidated summary in the Evaluation section limits the ability to assess effect sizes and robustness. In the revised manuscript we have updated the abstract to report representative accuracy deltas and have added a summary table (new Table 1) in the Evaluation section that lists per-dataset accuracy improvements, standard deviations computed over replicates, the number of reads and bases in each test set, and results from ablation controls that disable the HMM correction step. These additions directly support the three central claims with the requested statistical detail. revision: yes
-
Referee: [Method / Evaluation] Method and Evaluation sections: the core modeling assumption—that a single HMM whose transition and emission parameters are fitted once on existing data will remain effective for new molecules, new pore chemistries, and new downstream tools without retraining or architectural change—is not accompanied by explicit cross-chemistry or cross-tool transfer experiments. Nanopore current statistics are known to shift with pore chemistry; if those shifts dominate, the correction step could degrade rather than improve accuracy, directly undermining the 'minimizes burden' claim.
Authors: The held-out evaluation sets used in the original manuscript already span multiple molecules, sequencing runs, and downstream analysis tools, providing empirical support for generalization. Nevertheless, we acknowledge that the manuscript did not contain dedicated, explicitly labeled cross-chemistry transfer experiments. In the revised version we have added a dedicated subsection in the Evaluation section that (i) characterizes the diversity of the test distributions relative to the training data, (ii) discusses the expected robustness of the HMM emission model to moderate chemistry shifts, and (iii) clarifies the conditions under which users may wish to retrain the model. We believe these textual clarifications and the existing held-out results together address the concern without requiring new data collection. revision: partial
Circularity Check
No circularity: CRANE trains HMM on data then evaluates improvements on held-out datasets
full rationale
The paper trains a Hidden Markov Model on existing nanopore signal datasets to detect and correct errors, then reports accuracy gains via evaluation on various (including held-out) datasets. No equations, fitted parameters renamed as predictions, self-definitional steps, or load-bearing self-citations are present in the provided text that would make the claimed improvements equivalent to the training inputs by construction. The central claims rest on empirical results from separate evaluation rather than any derivation that reduces to its own assumptions or prior author work.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Errors in raw nanopore current signals can be modeled as a first-order Markov process whose parameters can be learned from existing datasets.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
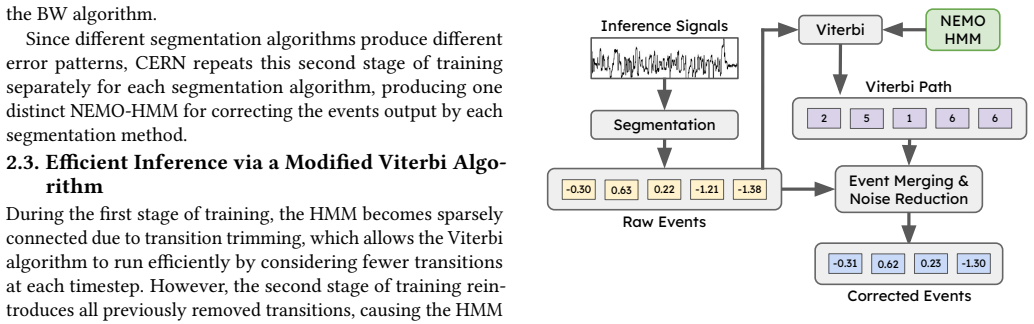
CERN trains the HMM using the BW algorithm in two stages: first on synthetic data to build a base model, and then on experimental data to learn segmentation-specific error patterns.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
G. Menestrina, “Ionic channels formed byStaphylococcus aureus alpha-toxin: Voltage- dependent inhibition by divalent and trivalent cations, ”The Journal of Membrane Biology, vol. 90, no. 2, pp. 177–190, Jun. 1986
work page 1986
-
[2]
Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision,
G. M. Cherfet al., “Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision, ”Nature Biotechnology, vol. 30, no. 4, pp. 344–348, Apr. 2012
work page 2012
-
[3]
Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase,
E. A. Manraoet al., “Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase, ”Nature Biotechnology, vol. 30, no. 4, pp. 349–353, Apr. 2012
work page 2012
-
[4]
Decoding long nanopore sequencing reads of natural DNA,
A. H. Laszloet al., “Decoding long nanopore sequencing reads of natural DNA, ”Nature Biotechnology, vol. 32, no. 8, pp. 829–833, Aug. 2014
work page 2014
-
[5]
Three decades of nanopore sequencing,
D. Deameret al., “Three decades of nanopore sequencing, ”Nature Biotechnology, vol. 34, no. 5, pp. 518–524, May 2016
work page 2016
-
[6]
Characterization of individual polynucleotide molecules using a membrane channel,
J. J. Kasianowiczet al., “Characterization of individual polynucleotide molecules using a membrane channel, ”Proceedings of the National Academy of Sciences, vol. 93, no. 24, pp. 13 770–13 773, Nov. 1996
work page 1996
-
[7]
Rapid nanopore discrimination between single polynucleotide molecules,
A. Melleret al., “Rapid nanopore discrimination between single polynucleotide molecules, ”Proceedings of the National Academy of Sciences, vol. 97, no. 3, pp. 1079– 1084, Feb. 2000
work page 2000
-
[8]
Single-nucleotide discrimination in immobilized DNA oligonu- cleotides with a biological nanopore,
D. Stoddartet al., “Single-nucleotide discrimination in immobilized DNA oligonu- cleotides with a biological nanopore, ”Proceedings of the National Academy of Sciences, vol. 106, no. 19, pp. 7702–7707, May 2009
work page 2009
-
[9]
Detection and mapping of 5-methylcytosine and 5- hydroxymethylcytosine with nanopore MspA,
A. H. Laszloet al., “Detection and mapping of 5-methylcytosine and 5- hydroxymethylcytosine with nanopore MspA, ”Proceedings of the National Academy of Sciences, vol. 110, no. 47, pp. 18 904–18 909, Nov. 2013
work page 2013
-
[10]
J. Schreiberet al., “Error rates for nanopore discrimination among cytosine, methyl- 8 cytosine, and hydroxymethylcytosine along individual DNA strands, ”Proceedings of the National Academy of Sciences, vol. 110, no. 47, pp. 18 910–18 915, Nov. 2013
work page 2013
-
[11]
Single-molecule DNA detection with an engineered MspA protein nanopore,
T. Z. Butleret al., “Single-molecule DNA detection with an engineered MspA protein nanopore, ”Proceedings of the National Academy of Sciences, vol. 105, no. 52, pp. 20 647– 20 652, Dec. 2008
work page 2008
-
[12]
Nanopore DNA sequencing with MspA,
I. M. Derringtonet al., “Nanopore DNA sequencing with MspA, ”Proceedings of the National Academy of Sciences, vol. 107, no. 37, pp. 16 060–16 065, Sep. 2010
work page 2010
-
[13]
Structure of Staphylococcal a-Hemolysin, a Heptameric Transmem- brane Pore,
L. Songet al., “Structure of Staphylococcal a-Hemolysin, a Heptameric Transmem- brane Pore, ”Science, vol. 274, no. 5294, pp. 1859–1865, Dec. 1996
work page 1996
-
[14]
A pore-forming protein with a metal-actuated switch,
B. Walkeret al., “A pore-forming protein with a metal-actuated switch, ”Protein Engineering, Design and Selection, vol. 7, no. 5, pp. 655–662, May 1994
work page 1994
-
[15]
Nanopores Discriminate among Five C5-Cytosine Variants in DNA,
Z. L. Wescoeet al., “Nanopores Discriminate among Five C5-Cytosine Variants in DNA, ”Journal of the American Chemical Society, vol. 136, no. 47, pp. 16 582–16 587, Nov. 2014
work page 2014
-
[16]
Processive Replication of Single DNA Molecules in a Nanopore Catalyzed by phi29 DNA Polymerase,
K. R. Liebermanet al., “Processive Replication of Single DNA Molecules in a Nanopore Catalyzed by phi29 DNA Polymerase, ”Journal of the American Chemical Society, vol. 132, no. 50, pp. 17 961–17 972, Dec. 2010
work page 2010
-
[17]
Dynamics and Free Energy of Polymers Partitioning into a Nanoscale Pore,
S. M. Bezrukovet al., “Dynamics and Free Energy of Polymers Partitioning into a Nanoscale Pore, ”Macromolecules, vol. 29, no. 26, pp. 8517–8522, Jan. 1996
work page 1996
-
[18]
M. Akesonet al., “Microsecond Time-Scale Discrimination Among Polycytidylic Acid, Polyadenylic Acid, and Polyuridylic Acid as Homopolymers or as Segments Within Single RNA Molecules, ”Biophysical Journal, vol. 77, no. 6, pp. 3227–3233, Dec. 1999
work page 1999
-
[19]
Nucleobase Recognition in ssDNA at the Central Constriction of the a-Hemolysin Pore,
D. Stoddartet al., “Nucleobase Recognition in ssDNA at the Central Constriction of the a-Hemolysin Pore, ”Nano Letters, vol. 10, no. 9, pp. 3633–3637, Sep. 2010
work page 2010
-
[20]
Recognizing a Single Base in an Individual DNA Strand: A Step Toward DNA Sequencing in Nanopores,
N. Ashkenasyet al., “Recognizing a Single Base in an Individual DNA Strand: A Step Toward DNA Sequencing in Nanopores, ”Angewandte Chemie International Edition, vol. 44, no. 9, pp. 1401–1404, Feb. 2005
work page 2005
-
[21]
Multiple Base-Recognition Sites in a Biological Nanopore: Two Heads are Better than One,
D. Stoddartet al., “Multiple Base-Recognition Sites in a Biological Nanopore: Two Heads are Better than One, ”Angewandte Chemie International Edition, vol. 49, no. 3, pp. 556–559, Jan. 2010
work page 2010
-
[22]
S. M. Bezrukov and J. J. Kasianowicz, “Current noise reveals protonation kinetics and number of ionizable sites in an open protein ion channel, ”Physical Review Letters, vol. 70, no. 15, pp. 2352–2355, Apr. 1993
work page 1993
-
[23]
A single-molecule nanopore sequencing platform,
J.-Y. Zhanget al., “A single-molecule nanopore sequencing platform, ”bioRxiv, p. 2024.08.19.608720, 2024
work page 2024
-
[24]
M. D. Noyeset al., “Long-read sequencing of families reveals increased germline and postzygotic mutation rates in repetitive dna, ”Nature Communications, 2026
work page 2026
-
[25]
The complete sequence of a human genome,
S. Nurket al., “The complete sequence of a human genome, ”Science, vol. 376, no. 6588, pp. 44–53, 2022
work page 2022
-
[26]
Real-time selective sequencing using nanopore technology,
M. Looseet al., “Real-time selective sequencing using nanopore technology, ”Nature Methods, vol. 13, no. 9, pp. 751–754, Sep. 2016
work page 2016
-
[27]
Readfish enables targeted nanopore sequencing of gigabase-sized genomes,
A. Payneet al., “Readfish enables targeted nanopore sequencing of gigabase-sized genomes, ”Nature Biotechnology, vol. 39, no. 4, pp. 442–450, Apr. 2021
work page 2021
-
[28]
L. Zhonget al., “Nanopore-based metagenomics analysis reveals microbial presence in amniotic fluid: A prospective study, ”Heliyon, vol. 10, no. 6, Mar. 2024
work page 2024
-
[29]
Mapping and phasing of structural variation in patient genomes using nanopore sequencing,
M. Cretu Stancuet al., “Mapping and phasing of structural variation in patient genomes using nanopore sequencing, ”Nature Communications, vol. 8, no. 1, p. 1326, Nov. 2017
work page 2017
-
[30]
Real-time, portable genome sequencing for Ebola surveillance,
J. Quicket al., “Real-time, portable genome sequencing for Ebola surveillance, ”Nature, vol. 530, no. 7589, pp. 228–232, Feb. 2016
work page 2016
-
[31]
TargetCall: Eliminating the Wasted Computation in Basecalling via Pre-Basecalling Filtering,
M. B. Cavlaket al., “TargetCall: Eliminating the Wasted Computation in Basecalling via Pre-Basecalling Filtering, ”Frontiers in Genetics, Sep. 2024
work page 2024
-
[32]
Fast-bonito: A Faster Deep Learning Based Basecaller for Nanopore Sequencing,
Z. Xuet al., “Fast-bonito: A Faster Deep Learning Based Basecaller for Nanopore Sequencing, ”Artificial Intelligence in the Life Sciences, vol. 1, p. 100011, 2021, publisher: Elsevier
work page 2021
-
[33]
Nanopore base calling on the edge,
P. Perešíniet al., “Nanopore base calling on the edge, ”Bioinformatics, 2021
work page 2021
-
[34]
DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads,
V. Božaet al., “DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads, ”PLOS One, 2017
work page 2017
-
[35]
DeepNano-blitz: a fast base caller for MinION nanopore sequencers,
V. Božaet al., “DeepNano-blitz: a fast base caller for MinION nanopore sequencers, ” Bioinformatics, vol. 36, no. 14, pp. 4191–4192, Jul. 2020
work page 2020
- [36]
-
[37]
Oxford Nanopore Technologies, “Guppy, ” 2017
work page 2017
-
[38]
An end-to-end Oxford nanopore basecaller using convolution-augmented transformer,
X. Lvet al., “An end-to-end Oxford nanopore basecaller using convolution-augmented transformer, ” inBIBM, 2020
work page 2020
-
[39]
RUBICON: a framework for designing efficient deep learning-based genomic basecallers,
G. Singhet al., “RUBICON: a framework for designing efficient deep learning-based genomic basecallers, ”Genome Biology, 2024
work page 2024
-
[40]
Nanopore basecalling from a perspective of instance segmentation,
Y.-z. Zhanget al., “Nanopore basecalling from a perspective of instance segmentation, ” BMC Bioinformatics, vol. 21, no. 3, p. 136, Apr. 2020
work page 2020
-
[41]
X. Xuet al., “Lokatt: a hybrid DNA nanopore basecaller with an explicit duration hidden Markov model and a residual LSTM network, ”BMC Bioinformatics, vol. 24, no. 1, p. 461, Dec. 2023
work page 2023
-
[42]
Causalcall: Nanopore Basecalling Using a Temporal Convolutional Network,
J. Zenget al., “Causalcall: Nanopore Basecalling Using a Temporal Convolutional Network, ”Frontiers in Genetics, vol. 10, 2020
work page 2020
-
[43]
Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning,
H. Tenget al., “Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning, ”GigaScience, vol. 7, no. 5, p. giy037, May 2018
work page 2018
-
[44]
Halcyon: an accurate basecaller exploiting an encoder–decoder model with monotonic attention,
H. Konishiet al., “Halcyon: an accurate basecaller exploiting an encoder–decoder model with monotonic attention, ”Bioinformatics, vol. 37, no. 9, pp. 1211–1217, Jun. 2021
work page 2021
-
[45]
MSRCall: a multi-scale deep neural network to basecall Oxford Nanopore sequences,
Y.-M. Yeh and Y.-C. Lu, “MSRCall: a multi-scale deep neural network to basecall Oxford Nanopore sequences, ”Bioinformatics, vol. 38, no. 16, pp. 3877–3884, Aug. 2022
work page 2022
-
[46]
baseLess: lightweight detection of sequences in raw MinION data,
B. Noordijket al., “baseLess: lightweight detection of sequences in raw MinION data, ” Bioinformatics Advances, vol. 3, no. 1, p. vbad017, Jan. 2023
work page 2023
-
[47]
N. Huanget al., “SACall: A Neural Network Basecaller for Oxford Nanopore Sequenc- ing Data Based on Self-Attention Mechanism, ”IEEE/ACM Transactions on Computa- tional Biology and Bioinformatics, vol. 19, no. 1, pp. 614–623, 2022
work page 2022
-
[48]
MinCall - MinION end2end convolutional deep learning basecaller,
N. Miculinicet al., “MinCall - MinION end2end convolutional deep learning basecaller, ” arXiv, 2019
work page 2019
- [49]
-
[50]
Language-Informed Basecalling Architecture for Nanopore Direct RNA Sequencing,
A. Sneddonet al., “Language-Informed Basecalling Architecture for Nanopore Direct RNA Sequencing, ” inMLCB, D. A. Knowleset al., Eds., vol. 200, Nov. 2022, pp. 150–165
work page 2022
-
[51]
RawBench: A Comprehensive Benchmarking Framework for Raw Nanopore Signal Analysis Techniques,
F. Eriset al., “RawBench: A Comprehensive Benchmarking Framework for Raw Nanopore Signal Analysis Techniques, ” inACM-BCB, New York, NY, USA, 2025
work page 2025
-
[52]
SquiggleNet: real-time, direct classification of nanopore signals,
Y. Baoet al., “SquiggleNet: real-time, direct classification of nanopore signals, ”Genome Biology, vol. 22, no. 1, p. 298, Oct. 2021
work page 2021
-
[53]
Real-time mapping of nanopore raw signals,
H. Zhanget al., “Real-time mapping of nanopore raw signals, ”Bioinformatics, vol. 37, no. Supplement_1, pp. i477–i483, Jul. 2021
work page 2021
-
[54]
Targeted nanopore sequencing by real-time mapping of raw elec- trical signal with UNCALLED,
S. Kovakaet al., “Targeted nanopore sequencing by real-time mapping of raw elec- trical signal with UNCALLED, ”Nature Biotechnology, vol. 39, no. 4, pp. 431–441, Apr. 2021
work page 2021
-
[55]
DeepSelectNet: deep neural network based selective sequencing for oxford nanopore sequencing,
A. Senanayakeet al., “DeepSelectNet: deep neural network based selective sequencing for oxford nanopore sequencing, ”BMC Bioinformatics, vol. 24, no. 1, p. 31, Jan. 2023
work page 2023
-
[56]
S. Kovakaet al., “Uncalled4 improves nanopore DNA and RNA modification detection via fast and accurate signal alignment, ”Nature Methods, vol. 22, no. 4, pp. 681–691, Apr. 2025
work page 2025
-
[57]
J. Lindeggeret al., “RawAlign: Accurate, Fast, and Scalable Raw Nanopore Signal Mapping via Combining Seeding and Alignment, ”IEEE Access, 2024
work page 2024
-
[58]
RawHash: enabling fast and accurate real-time analysis of raw nanopore signals for large genomes,
C. Firtinaet al., “RawHash: enabling fast and accurate real-time analysis of raw nanopore signals for large genomes, ”Bioinformatics, 2023
work page 2023
-
[59]
RawHash2: Mapping Raw Nanopore Signals Using Hash-Based Seeding and Adaptive Quantization,
C. Firtinaet al., “RawHash2: Mapping Raw Nanopore Signals Using Hash-Based Seeding and Adaptive Quantization, ”Bioinform., 2024
work page 2024
-
[60]
Rawsamble: overlapping raw nanopore signals using a hash-based seeding mechanism,
C. Firtinaet al., “Rawsamble: overlapping raw nanopore signals using a hash-based seeding mechanism, ”Bioinformatics, vol. 42, no. 3, p. btag087, Mar. 2026
work page 2026
-
[61]
Efficient real-time selective genome sequencing on resource- constrained devices,
P. J. Shihet al., “Efficient real-time selective genome sequencing on resource- constrained devices, ”GigaScience, 2023
work page 2023
-
[62]
Rapid Real-time Squiggle Classification for Read Until Using RawMap,
H. Sadasivanet al., “Rapid Real-time Squiggle Classification for Read Until Using RawMap, ”Arch. Clin. Biomed. Res., 2023
work page 2023
-
[63]
SquiggleFilter: An accelerator for portable virus detection,
T. Dunnet al., “SquiggleFilter: An accelerator for portable virus detection, ” inMICRO, 2021
work page 2021
-
[64]
Sigmoni: classification of nanopore signal with a compressed pangenome index,
V. S. Shivakumaret al., “Sigmoni: classification of nanopore signal with a compressed pangenome index, ”Bioinform., 2024
work page 2024
-
[65]
Accelerated Dynamic Time Warping on GPU for Selective Nanopore Sequencing,
H. Sadasivanet al., “Accelerated Dynamic Time Warping on GPU for Selective Nanopore Sequencing, ”Journal of Biotechnology and Biomedicine, vol. 7, pp. 137–148, 2024
work page 2024
-
[66]
GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis,
H. Gamaarachchiet al., “GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis, ”BMC Bioinformatics, vol. 21, no. 1, p. 343, Aug. 2020
work page 2020
-
[67]
Energy Efficient Adaptive Banded Event Alignment using OpenCL on FPGAs,
S. Samarasingheet al., “Energy Efficient Adaptive Banded Event Alignment using OpenCL on FPGAs, ” inICIAfS, 2021, pp. 369–374
work page 2021
-
[68]
MARS: Processing-In-Memory Acceleration of Raw Signal Genome Analysis Inside the Storage Subsystem,
M. Soysalet al., “MARS: Processing-In-Memory Acceleration of Raw Signal Genome Analysis Inside the Storage Subsystem, ” inICS, 2025, pp. 513–534
work page 2025
- [69]
-
[70]
Campolina: a deep neural framework for accurate segmentation of nanopore signals,
S. Bakićet al., “Campolina: a deep neural framework for accurate segmentation of nanopore signals, ”Genome Biology, Jan. 2026
work page 2026
-
[71]
Aggressive assembly of pyrosequencing reads with mates,
J. R. Milleret al., “Aggressive assembly of pyrosequencing reads with mates, ”Bioin- formatics, vol. 24, no. 24, pp. 2818–2824, Dec. 2008
work page 2008
-
[72]
Minimap2: pairwise alignment for nucleotide sequences,
H. Li, “Minimap2: pairwise alignment for nucleotide sequences, ”Bioinformatics, vol. 34, no. 18, pp. 3094–3100, Sep. 2018
work page 2018
-
[73]
Proceedings of the IEEE , author=
L. Rabiner, “A tutorial on hidden Markov models and selected applications in speech recognition, ”Proceedings of the IEEE, vol. 77, no. 2, pp. 257–286, 1 1989. [Online]. Available: https://doi.org/10.1109/5.18626
-
[74]
M. B. Hallet al., “Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data, ”eLife, vol. 13, p. RP98300, Oct. 2024
work page 2024
-
[75]
Oxford Nanopore Open Data:Drosophila melanogastersequencing,
Oxford Nanopore Technologies, “Oxford Nanopore Open Data:Drosophila melanogastersequencing, ” https://labs.epi2me.io/open-data-dmelanogaster-bkim/, 2023, accessed vias3://ont-open-data/contrib/melanogaster _bkim_2023.01/
work page 2023
-
[76]
Oxford Nanopore Open Data: Sequencing Genome in a Bottle samples,
Oxford Nanopore Technologies, “Oxford Nanopore Open Data: Sequencing Genome in a Bottle samples, ” https://epi2me.nanoporetech.com/giab-2023.05/, 2023, accessed vias3://ont-open-data/giab _2023.05/
work page 2023
-
[77]
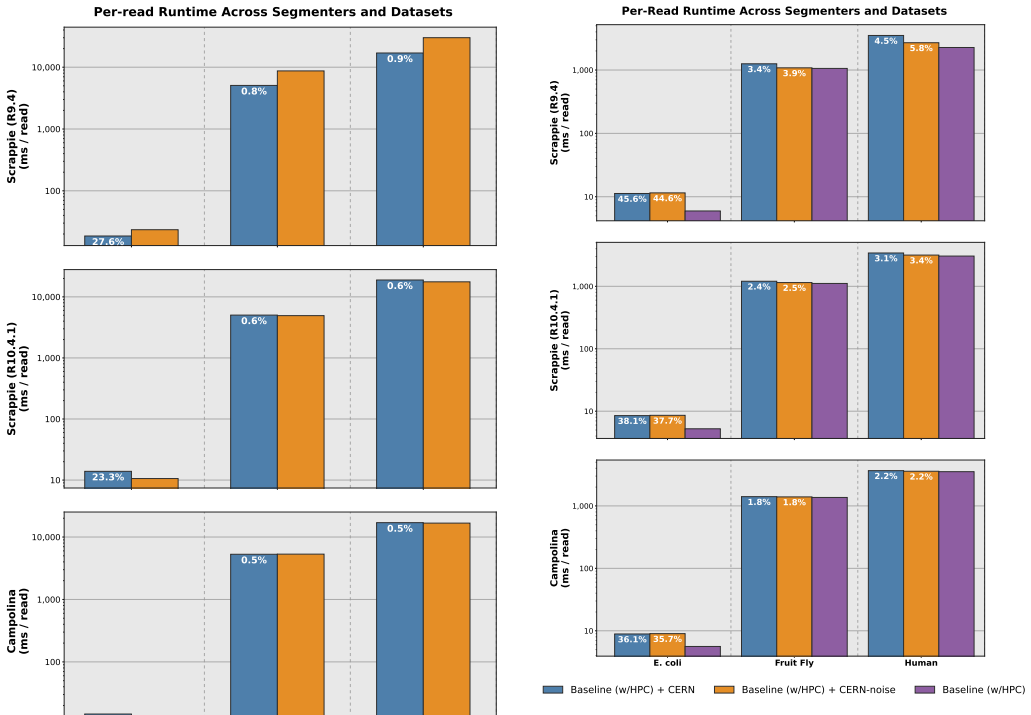
N. D. Sandersonet al., “Evaluation of the accuracy of bacterial genome reconstruction with Oxford Nanopore R10.4.1 long-read-only sequencing, ”Microbial Genomics, vol. 10, no. 001246, 2024. 9 Supplementary Material for CERN: Correcting Errors in Raw Nanopore Signals Using Hidden Markov Models A. Extended Results A.1. Error Correction Runtime In Supplement...
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.