Recognition: unknown
bioLeak: Leakage-Aware Modeling and Diagnostics for Machine Learning in R
Pith reviewed 2026-05-10 16:25 UTC · model grok-4.3
The pith
An R package called bioLeak constructs leakage-aware data splits and audits fitted models to reduce optimistic bias in biomedical machine learning.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
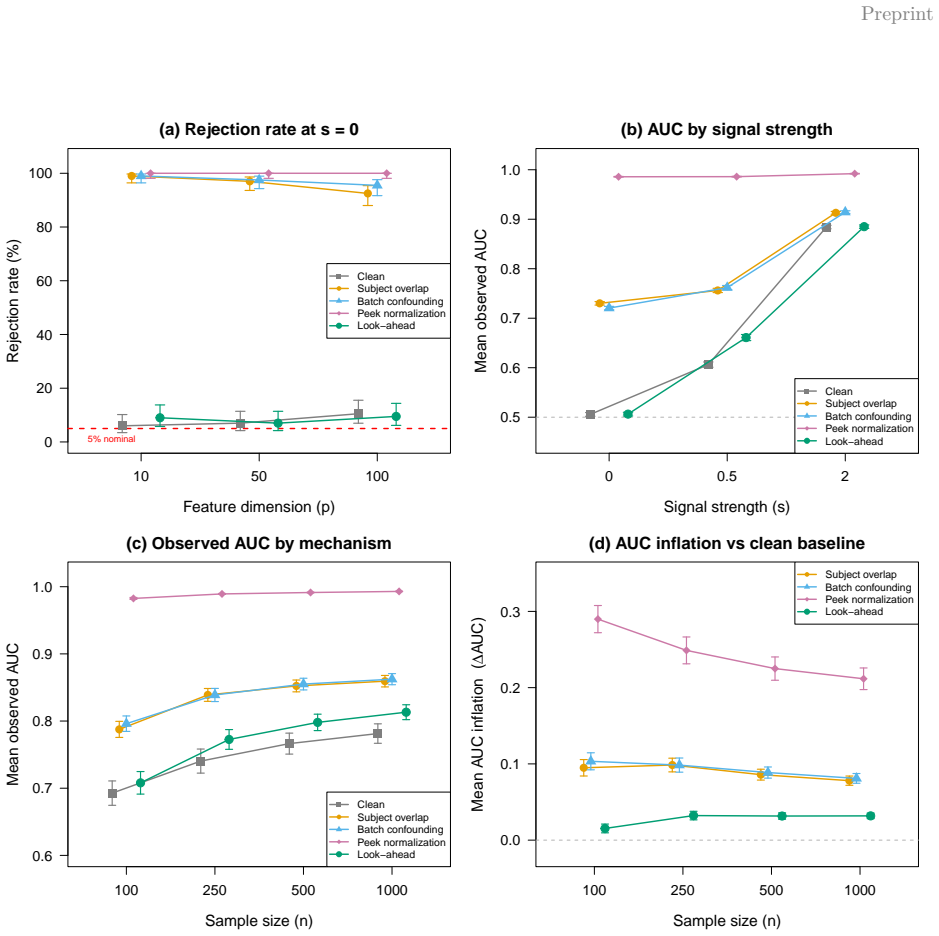
The package provides tools for leakage-aware split construction, train-fold-only preprocessing, cross-validated model fitting, nested hyperparameter tuning, post hoc leakage audits, and HTML reporting. Simulations show how apparent performance changes under controlled leakage, and the case study shows guarded and leaky pipelines yield materially different conclusions on multi-study transcriptomic data.
What carries the argument
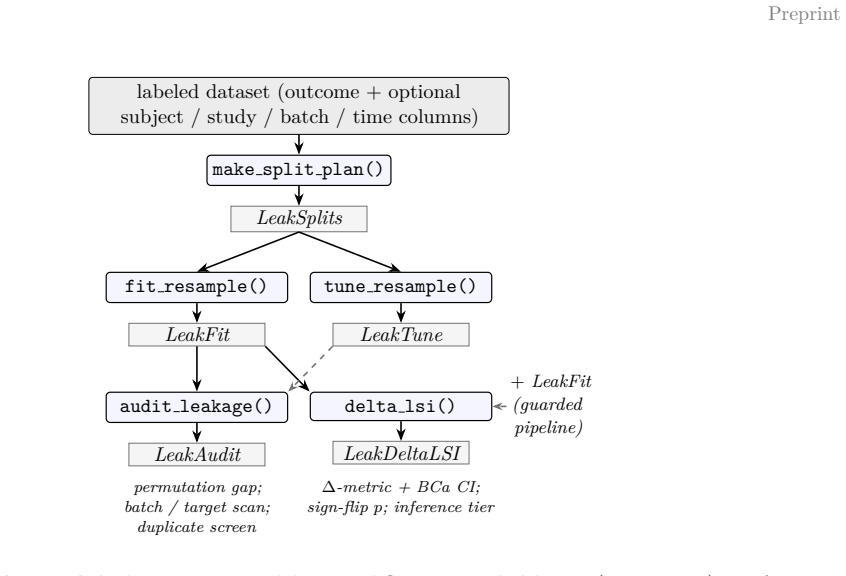
Leakage-aware split construction, train-fold-only preprocessing, and post-hoc leakage audits with S4 containers for splits, fits, audits, and inflation summaries.
If this is right
- Apparent performance decreases when leakage is prevented in simulations.
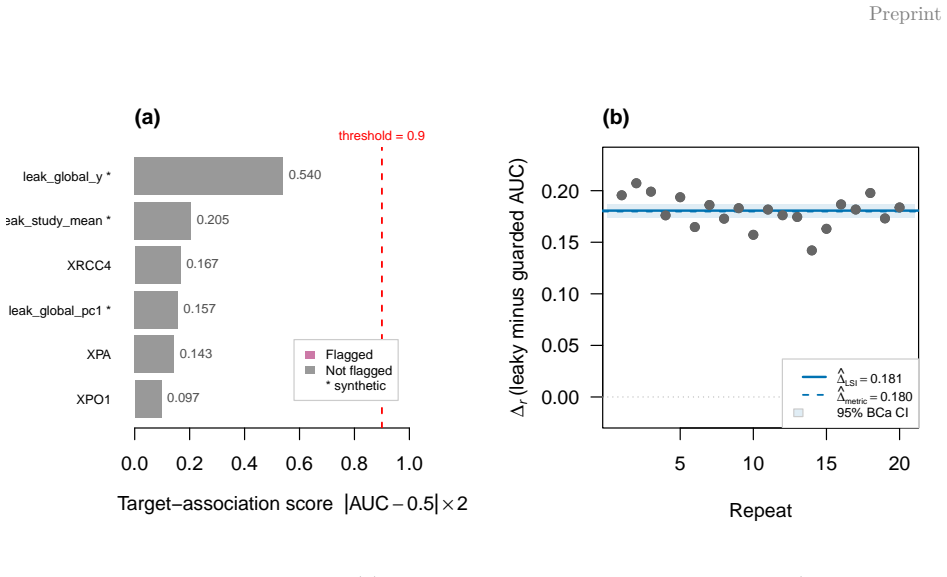
- Guarded pipelines can yield different conclusions than leaky ones in transcriptomic data.
- HTML reports aid interpretation of diagnostic output.
- The package supports binary and multiclass classification, regression, and survival analysis.
Where Pith is reading between the lines
- This software could help standardize leakage prevention in biomedical ML research.
- It might be extended to handle more complex data dependencies like temporal structures.
- Wider adoption could improve the reliability of published ML results in the field.
- Testing on datasets from other domains would verify generalizability.
Load-bearing premise
The package correctly identifies and blocks all relevant leakage mechanisms in typical biomedical datasets without introducing new biases.
What would settle it
A controlled experiment where a known leakage mechanism is not detected by the audits but still inflates performance would falsify the effectiveness of the diagnostics.
Figures

read the original abstract
Data leakage remains a recurrent source of optimistic bias in biomedical machine learning studies. Standard row-wise cross-validation and globally estimated preprocessing steps are often inappropriate for data with repeated measurements, study-level heterogeneity, batch effects, or temporal dependencies. This paper describes bioLeak, an R package for constructing leakage-aware resampling workflows and for auditing fitted models for common leakage mechanisms. The package provides leakage-aware split construction, train-fold-only preprocessing, cross-validated model fitting, nested hyperparameter tuning, post hoc leakage audits, and HTML reporting. The implementation supports binary classification, multiclass classification, regression, and survival analysis, with task-specific metrics and S4 containers for splits, fits, audits, and inflation summaries. The simulation artifacts show how apparent performance changes under controlled leakage mechanisms, and the case study illustrates how guarded and leaky pipelines can yield materially different conclusions on multi-study transcriptomic data. The emphasis throughout is on software design, reproducible workflows, and interpretation of diagnostic output.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript describes bioLeak, an R package for constructing leakage-aware resampling workflows and auditing fitted models for common leakage mechanisms in biomedical ML. It supports binary/multiclass classification, regression, and survival analysis via leakage-aware splits, train-fold-only preprocessing, nested tuning, post-hoc audits, and HTML reporting. Simulations illustrate performance changes under controlled leakage, and a case study on multi-study transcriptomic data shows differing conclusions between guarded and leaky pipelines.
Significance. If the audits and workflows perform as intended, the package addresses a key source of optimistic bias in biomedical ML, promoting more reliable analyses through reproducible, task-specific tools. Credit is due for the S4 container design, broad task support, and focus on diagnostic interpretation and workflow reproducibility.
major comments (2)
- [Simulations] The abstract and simulation description claim that artifacts show how apparent performance changes under controlled leakage mechanisms, but no quantitative results (e.g., specific metrics like AUC or RMSE, sample sizes, or comparisons with/without error bars) are reported; this undermines the illustrative evidence for the package's value (§ Simulations).
- [Case study] The case study claims that guarded and leaky pipelines yield materially different conclusions on transcriptomic data, yet provides no details on the exact datasets, models, metrics, or statistical criteria used to establish 'material' difference; this is load-bearing for demonstrating practical impact (§ Case study).
minor comments (2)
- The manuscript would benefit from explicit installation instructions, a minimal reproducible example, and a table summarizing supported task types and metrics.
- [Methods] Notation for S4 classes (splits, fits, audits) could be introduced with a small diagram or example output to improve accessibility for biomedical users.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review of our manuscript on the bioLeak R package. We address each major comment below and will incorporate revisions to strengthen the quantitative support in the simulations and case study sections.
read point-by-point responses
-
Referee: [Simulations] The abstract and simulation description claim that artifacts show how apparent performance changes under controlled leakage mechanisms, but no quantitative results (e.g., specific metrics like AUC or RMSE, sample sizes, or comparisons with/without error bars) are reported; this undermines the illustrative evidence for the package's value (§ Simulations).
Authors: We agree that the simulations section would be strengthened by explicit quantitative reporting in the manuscript text. Although the simulation artifacts (figures and reproducible code) illustrate performance shifts under controlled leakage, specific metrics such as AUC or RMSE, sample sizes, and error-bar comparisons are not detailed in the current text. In the revised manuscript we will add these quantitative results, including example values and direct with/without-leakage comparisons, to better substantiate the package's value. revision: yes
-
Referee: [Case study] The case study claims that guarded and leaky pipelines yield materially different conclusions on transcriptomic data, yet provides no details on the exact datasets, models, metrics, or statistical criteria used to establish 'material' difference; this is load-bearing for demonstrating practical impact (§ Case study).
Authors: We acknowledge that the case study lacks the necessary specifics to fully support the claim of materially different conclusions. The manuscript currently refers to multi-study transcriptomic data without enumerating datasets, models, metrics, or the criteria for 'material' difference. In revision we will expand this section to specify the exact datasets (including sources and sample sizes), models fitted, performance metrics used, and the statistical or practical thresholds applied to establish differing conclusions between guarded and leaky pipelines. revision: yes
Circularity Check
No significant circularity; software description without derivation chain
full rationale
The manuscript is a software package description (bioLeak in R) focused on implementing standard leakage-aware resampling, train-only preprocessing, nested tuning, and post-hoc audits. No equations, fitted parameters, or mathematical derivations are presented that could reduce to their own inputs by construction. Simulations and the transcriptomic case study serve as illustrations of performance differences rather than load-bearing claims derived from self-citations or ansatzes. No self-definitional, fitted-input, or uniqueness-imported steps exist; the work is self-contained as a tool for reproducible workflows.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Karthik Ram, Carl Boettiger, Scott Chamberlain, Noam Ross, Maelle Goldberg, and Ignasi Bartomeus
doi: 10.1126/science.1213847. 33 Preprint Belinda Phipson and Gordon K. Smyth. Permutation p-values should never be zero: Calculating exact p-values when permutations are randomly drawn.Statistical Applications in Genetics and Molecular Biology, 9(1):Article 39, 2010. doi: 10.2202/1544-6115.1585. R Core Team.R: A Language and Environment for Statistical C...
-
[2]
and Bahn, Volker and Ciuti, Simone and Boyce, Mark S
doi: 10.1111/ecog.02881. Jonathan D. Rosenblatt, Yaron Ben Simhon, Sagiv Gal, and Ro’ee Gilron. Practicalities of resampling methods for brain imaging: What every researcher should know.NeuroImage, 287:120520, 2024. doi: 10.1016/j.neuroimage.2024.120520. Geir Kjetil Sandve, Anton Nekrutenko, James Taylor, and Eivind Hovig. Ten simple rules for reproducibl...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.