Recognition: unknown
Phylogenetic Inference under the Balanced Minimum Evolution Criterion via Semidefinite Programming
Pith reviewed 2026-05-10 14:36 UTC · model grok-4.3
The pith
Semidefinite programming relaxes the balanced minimum evolution problem to recover accurate phylogenetic trees.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
An SDP relaxation of the BME objective, combined with an iterative rounding scheme, converts relaxed matrix solutions into valid tree topologies and produces accurate phylogenetic reconstructions on both simulated and empirical data.
What carries the argument
The SDP relaxation of the balanced minimum evolution objective function together with an iterative rounding procedure that maps positive-semidefinite matrices back to tree topologies.
If this is right
- The SDP-plus-rounding pipeline reconstructs phylogenies accurately on both simulated and real data.
- The same convex-relaxation strategy can be extended to other phylogenetic optimization problems.
- Distance-based inference gains access to the same tight convex bounds already used in many combinatorial settings outside biology.
- Larger or noisier distance matrices become tractable once the problem is moved into the positive-semidefinite cone.
Where Pith is reading between the lines
- If the rounding step proves stable, the framework could replace heuristic search in pipelines that currently rely on neighbor-joining or fastME variants.
- The approach invites direct comparison with integer-programming formulations of the same BME objective to quantify the price of the convex relaxation.
- Because SDP solvers scale with matrix size rather than with the number of tree topologies, the method may open the door to inference on hundreds of taxa where exhaustive search remains impossible.
Load-bearing premise
The SDP relaxation of the BME objective stays sufficiently tight that the rounding procedure recovers high-quality or optimal trees without frequent failure or prohibitive cost.
What would settle it
On standard benchmark datasets the method would return trees whose Robinson-Foulds distance to the true topology exceeds the distance achieved by established BME heuristics.
Figures

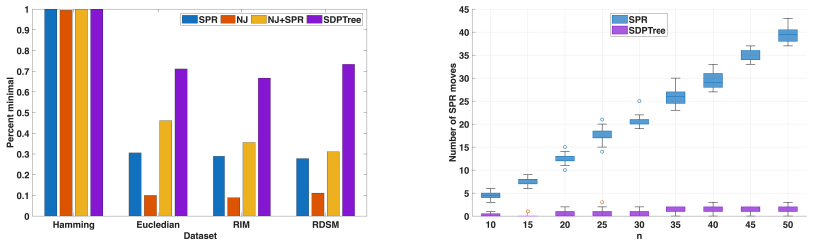
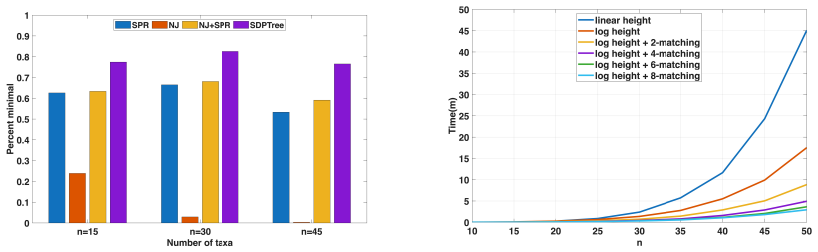
read the original abstract
In this study, we investigate the application of Semidefinite Programming (SDP) to phylogenetics. SDP is a powerful optimization framework that seeks to optimize a linear objective function over the cone of positive semidefinite matrices. As a convex optimization problem, SDP generalizes linear programming and provides tight relaxations for many combinatorial optimization problems. However, despite its many applications, SDP remains largely unused in computational biology. We argue that SDP relaxations are particularly well suited for phylogenetic inference. As a proof of concept, we focus on the Balanced Minimum Evolution (BME) problem, a widely used model in distance-based phylogenetics. We propose an algorithm combining an SDP relaxation with a rounding scheme that iteratively converts relaxed solutions into valid tree topologies. Experiments on simulated and empirical datasets show that the method enables accurate phylogenetic reconstruction. The approach is sufficiently general to be extendable to other phylogenetic problems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an SDP relaxation of the Balanced Minimum Evolution (BME) objective for distance-based phylogenetic inference, combined with a deterministic iterative rounding procedure that projects relaxed solutions onto valid tree topologies. Experiments on simulated data (recovery of known topologies) and empirical datasets (comparisons to NJ and other methods) report Robinson-Foulds distances and runtimes that scale to a few dozen taxa, with the claim that the framework is extensible to other phylogenetic problems.
Significance. If the reported performance holds, the work supplies a convex-optimization route to a standard phylogenetic criterion that is distinct from existing heuristics and admits a standard SDP formulation whose tightness is directly tested by the rounding success rate. The explicit reporting of RF distances, runtime scaling, and deterministic rounding constitute reproducible evidence that addresses the central practical question of whether the relaxation remains useful after projection.
minor comments (3)
- [Abstract] Abstract: the claim of 'accurate phylogenetic reconstruction' is not accompanied by any numerical summary (e.g., mean RF distance or success rate); a single sentence with the key quantitative result would make the abstract self-contained.
- [Methods] The description of the iterative rounding scheme (projection steps, termination criterion, and handling of numerical tolerance) is only sketched; a short pseudocode block or explicit list of the projection operations would remove ambiguity for readers wishing to re-implement the method.
- [Experiments] Table or figure captions for the empirical results should explicitly state the number of taxa, number of replicates, and the precise distance matrix source for each dataset so that the reported RF values can be directly compared with future work.
Simulated Author's Rebuttal
We thank the referee for their positive summary of our work, recognition of its significance as a convex-optimization approach to BME, and recommendation for minor revision. The referee's description accurately reflects the SDP relaxation, deterministic iterative rounding, and the reported experiments on simulated and empirical data.
Circularity Check
No significant circularity
full rationale
The paper formulates an SDP relaxation of the standard BME objective (linear function over PSD matrices with tree topology constraints) and applies a deterministic iterative rounding procedure to recover topologies. This follows conventional convex optimization techniques with no reduction of outputs to fitted inputs, self-definitional equations, or load-bearing self-citations. Validation on simulated and empirical data (Robinson-Foulds distances, runtime scaling) is independent of the formulation itself, confirming the derivation chain is self-contained.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Determining protein structures from noesy distance constraints by semidefinite programming
Babak Alipanahi, Nathan Krislock, Ali Ghodsi, Henry Wolkowicz, Logan Donaldson, and Ming Li. Determining protein structures from noesy distance constraints by semidefinite programming. Journal of Computational Biology, 20(4):296–310, 2013
2013
-
[2]
The performance of neighbor-joining methods of phylogenetic reconstruction
Kevin Atteson. The performance of neighbor-joining methods of phylogenetic reconstruction. Algorithmica, 25(2):251–278, 1999
1999
-
[3]
SIAM, 2001
Aharon Ben-Tal and Arkadi Nemirovski.Lectures on modern convex optimization: analysis, algorithms, and engineering applications. SIAM, 2001
2001
-
[4]
Cambridge university press, 2004
Stephen Boyd, Stephen P Boyd, and Lieven Vandenberghe.Convex optimization. Cambridge university press, 2004
2004
-
[5]
A method for deducing branching sequences in phylogeny
Joseph H Camin and Robert R Sokal. A method for deducing branching sequences in phylogeny. Evolution, 19(3):311–326, September 1965
1965
-
[6]
New heuristics for phylogeny estimation under the balanced minimum evolution criterion.Bioinformatics, 41(7):btaf361, 2025
Daniele Catanzaro, Henri Dehaybe, and Raffaele Pesenti. New heuristics for phylogeny estimation under the balanced minimum evolution criterion.Bioinformatics, 41(7):btaf361, 2025
2025
-
[7]
The bal- anced minimum evolution problem.INFORMS Journal on Computing, 24(2):276–294, 2012
Daniele Catanzaro, Martine Labb´ e, Raffaele Pesenti, and Juan-Jos´ e Salazar-Gonz´ alez. The bal- anced minimum evolution problem.INFORMS Journal on Computing, 24(2):276–294, 2012
2012
-
[8]
Optimizing over path-length matrices of unrooted binary trees.Mathematical Programming, 215(1):453–505, 2026
Daniele Catanzaro, Raffaele Pesenti, Allan Sapucaia, and Laurence Wolsey. Optimizing over path-length matrices of unrooted binary trees.Mathematical Programming, 215(1):453–505, 2026
2026
-
[9]
On the balanced minimum evolution polytope.Discrete Optimization, 36:100570, 2020
Daniele Catanzaro, Raffaele Pesenti, and Laurence Wolsey. On the balanced minimum evolution polytope.Discrete Optimization, 36:100570, 2020
2020
-
[10]
Maximum likelihood of evolutionary trees: hardness and approxi- mation.Bioinformatics, 21(suppl 1):i97–i106, 2005
Benny Chor and Tamir Tuller. Maximum likelihood of evolutionary trees: hardness and approxi- mation.Bioinformatics, 21(suppl 1):i97–i106, 2005
2005
-
[11]
John Wiley & Sons, 1999
Thomas M Cover.Elements of information theory. John Wiley & Sons, 1999
1999
-
[12]
Computational complexity of inferring phylogenies from dissimilarity matrices
William HE Day. Computational complexity of inferring phylogenies from dissimilarity matrices. Bulletin of mathematical biology, 49(4):461–467, 1987
1987
-
[13]
Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle.Journal of computational biology, 9(5):687–705, 2002
Richard Desper and Olivier Gascuel. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle.Journal of computational biology, 9(5):687–705, 2002
2002
-
[14]
Theoretical foundation of the balanced minimum evolu- tion method of phylogenetic inference and its relationship to weighted least-squares tree fitting
Richard Desper and Olivier Gascuel. Theoretical foundation of the balanced minimum evolu- tion method of phylogenetic inference and its relationship to weighted least-squares tree fitting. Molecular Biology and Evolution, 21(3):587–598, 2004
2004
-
[15]
Benchmarking optimization software with performance profiles.Mathematical programming, 91(2):201–213, 2002
Elizabeth D Dolan and Jorge J Mor´ e. Benchmarking optimization software with performance profiles.Mathematical programming, 91(2):201–213, 2002
2002
-
[16]
Origin and evolution of the octoploid strawberry genome.Nature genetics, 51(3):541–547, 2019
Patrick P Edger, Thomas J Poorten, Robert VanBuren, Michael A Hardigan, Marivi Colle, Michael R McKain, Ronald D Smith, Scott J Teresi, Andrew DL Nelson, Ching Man Wai, et al. Origin and evolution of the octoploid strawberry genome.Nature genetics, 51(3):541–547, 2019
2019
-
[17]
Convergence assessment for bayesian phylogenetic analysis using mcmc simulation.Methods in Ecology and Evolution, 13(1):77–90, 2022
Luiza Guimaraes Fabreti and Sebastian H¨ ohna. Convergence assessment for bayesian phylogenetic analysis using mcmc simulation.Methods in Ecology and Evolution, 13(1):77–90, 2022
2022
-
[18]
Approximating the balanced minimum evolution problem
Samuel Fiorini and Gwena¨ el Joret. Approximating the balanced minimum evolution problem. Operations research letters, 40(1):31–35, 2012
2012
-
[19]
Facets of the balanced minimal evolution poly- tope.Journal of mathematical biology, 73:447–468, 2016
Stefan Forcey, Logan Keefe, and William Sands. Facets of the balanced minimal evolution poly- tope.Journal of mathematical biology, 73:447–468, 2016. 10
2016
-
[20]
On the approximability of the fixed-tree balanced minimum evolution problem
Martin Frohn. On the approximability of the fixed-tree balanced minimum evolution problem. Optimization Letters, 15(6):2321–2329, 2021
2021
-
[21]
Neighbor-joining revealed.Molecular biology and evolution, 23(11):1997–2000, 2006
Olivier Gascuel and Mike Steel. Neighbor-joining revealed.Molecular biology and evolution, 23(11):1997–2000, 2006
1997
-
[22]
An evolution strategy approach for the balanced minimum evolution problem.Bioinformatics, 39(11):btad660, 2023
Andrea Gasparin, Federico Julian Camerota Verd` u, Daniele Catanzaro, and Lorenzo Castelli. An evolution strategy approach for the balanced minimum evolution problem.Bioinformatics, 39(11):btad660, 2023
2023
-
[23]
Improved approximation algorithms for maximum cut and satisfiability problems using semidefinite programming.Journal of the ACM (JACM), 42(6):1115–1145, 1995
Michel X Goemans and David P Williamson. Improved approximation algorithms for maximum cut and satisfiability problems using semidefinite programming.Journal of the ACM (JACM), 42(6):1115–1145, 1995
1995
-
[24]
Cambridge University Press, 2019
Dan Gusfield.Integer linear programming in computational and systems biology. Cambridge University Press, 2019
2019
-
[25]
Haplotype phasing using semidefinite program- ming
Konstantinos Kalpakis and Parag Namjoshi. Haplotype phasing using semidefinite program- ming. InFifth IEEE Symposium on Bioinformatics and Bioengineering (BIBE’05), pages 145–152. IEEE, 2005
2005
-
[26]
On the genealogy of large populations.Journal of applied probability, 19(A):27– 43, 1982
John FC Kingman. On the genealogy of large populations.Journal of applied probability, 19(A):27– 43, 1982
1982
-
[27]
Cellphy: accurate and fast probabilistic inference of single-cell phylogenies from scdna-seq data.Genome biology, 23(1):37, 2022
Alexey Kozlov, Joao M Alves, Alexandros Stamatakis, and David Posada. Cellphy: accurate and fast probabilistic inference of single-cell phylogenies from scdna-seq data.Genome biology, 23(1):37, 2022
2022
-
[28]
Fastme 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program.Molecular biology and evolution, 32(10):2798– 2800, 2015
Vincent Lefort, Richard Desper, and Olivier Gascuel. Fastme 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program.Molecular biology and evolution, 32(10):2798– 2800, 2015
2015
-
[29]
The origins and spread of domestic horses from the western eurasian steppes.Nature, 598(7882):634– 640, 2021
Pablo Librado, Naveed Khan, Antoine Fages, Mariya A Kusliy, Tomasz Suchan, Laure Tonasso- Calvi` ere, St´ ephanie Schiavinato, Duha Alioglu, Aurore Fromentier, Aude Perdereau, et al. The origins and spread of domestic horses from the western eurasian steppes.Nature, 598(7882):634– 640, 2021
2021
-
[30]
High accuracy meets high throughput for near full-length 16s ribosomal rna amplicon sequencing on the nanopore platform.PNAS nexus, 3(10):pgae411, 2024
Xuan Lin, Katherine Waring, Hans Ghezzi, Carolina Tropini, John Tyson, and Ryan M Ziels. High accuracy meets high throughput for near full-length 16s ribosomal rna amplicon sequencing on the nanopore platform.PNAS nexus, 3(10):pgae411, 2024
2024
-
[31]
The influence of the number of tree searches on maximum likelihood inference in phylogenomics.Systematic Biology, 73(5):807–822, 2024
Chao Liu, Xiaofan Zhou, Yuanning Li, Chris Todd Hittinger, Ronghui Pan, Jinyan Huang, Xue- xin Chen, Antonis Rokas, Yun Chen, and Xing-Xing Shen. The influence of the number of tree searches on maximum likelihood inference in phylogenomics.Systematic Biology, 73(5):807–822, 2024
2024
-
[32]
L¨ ofberg
J. L¨ ofberg. Yalmip : A toolbox for modeling and optimization in matlab. InIn Proceedings of the CACSD Conference, Taipei, Taiwan, 2004
2004
-
[33]
Semidefinite programs and combinatorial optimization
L´ aszl´ o Lov´ asz. Semidefinite programs and combinatorial optimization. InRecent advances in algorithms and combinatorics, pages 137–194. Springer, 2003
2003
-
[34]
Global optimization in protein docking using clustering, underestimation and semidefinite programming.Optimisation Methods and Software, 22(5):803–811, 2007
Roummel F Marcia, Julie C Mitchell, and Stephen J Wright. Global optimization in protein docking using clustering, underestimation and semidefinite programming.Optimisation Methods and Software, 22(5):803–811, 2007
2007
-
[35]
Computability of global solutions to factorable nonconvex programs: Part i—convex underestimating problems.Mathematical programming, 10(1):147–175, 1976
Garth P McCormick. Computability of global solutions to factorable nonconvex programs: Part i—convex underestimating problems.Mathematical programming, 10(1):147–175, 1976
1976
-
[36]
Direct calculation of a tree length using a distance matrix.Journal of Molecular Evolution, 51:41–47, 2000
Yves Pauplin. Direct calculation of a tree length using a distance matrix.Journal of Molecular Evolution, 51:41–47, 2000. 11
2000
-
[37]
The neighbor-joining method: a new method for reconstructing phylogenetic trees.Mol
N Saitou and M Nei. The neighbor-joining method: a new method for reconstructing phylogenetic trees.Mol. Biol. Evol., 4(4):406–425, 1987
1987
-
[38]
Cyclic permutations and evolutionary trees.Advances in Applied Mathematics, 32(4):669–680, 2004
Charles Semple and Mike Steel. Cyclic permutations and evolutionary trees.Advances in Applied Mathematics, 32(4):669–680, 2004
2004
-
[39]
IOP Publishing, 2023
Paul Skrzypczyk and Daniel Cavalcanti.Semidefinite programming in quantum information sci- ence. IOP Publishing, 2023
2023
-
[40]
Phy- lobench: A benchmark for evaluating phylogenetic programs.Molecular Biology and Evolution, 41(6):msae084, 2024
Sergey Spirin, Andrey Sigorskikh, Aleksei Efremov, Dmitry Penzar, and Anna Karyagina. Phy- lobench: A benchmark for evaluating phylogenetic programs.Molecular Biology and Evolution, 41(6):msae084, 2024
2024
-
[41]
Semidefinite programming.SIAM review, 38(1):49–95, 1996
Lieven Vandenberghe and Stephen Boyd. Semidefinite programming.SIAM review, 38(1):49–95, 1996
1996
-
[42]
Trycycler: consensus long-read assemblies for bacterial genomes.Genome biology, 22(1):266, 2021
Ryan R Wick, Louise M Judd, Louise T Cerdeira, Jane Hawkey, Guillaume M´ eric, Ben Vezina, Kelly L Wyres, and Kathryn E Holt. Trycycler: consensus long-read assemblies for bacterial genomes.Genome biology, 22(1):266, 2021
2021
-
[43]
Springer Science & Business Media, 2012
Henry Wolkowicz, Romesh Saigal, and Lieven Vandenberghe.Handbook of semidefinite program- ming: theory, algorithms, and applications. Springer Science & Business Media, 2012
2012
-
[44]
Evaluating fast maxi- mum likelihood-based phylogenetic programs using empirical phylogenomic data sets.Molecular biology and evolution, 35(2):486–503, 2018
Xiaofan Zhou, Xing-Xing Shen, Chris Todd Hittinger, and Antonis Rokas. Evaluating fast maxi- mum likelihood-based phylogenetic programs using empirical phylogenomic data sets.Molecular biology and evolution, 35(2):486–503, 2018. 12
2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.