Recognition: unknown
Enhancing Cryo-EM Density Map Segmentation in Phenix for Improved Atomic Model Building
Pith reviewed 2026-05-09 16:40 UTC · model grok-4.3
The pith
PhenixCraft integrates AlphaFold predictions to improve cryo-EM density map segmentation in Phenix, yielding higher TM-scores and sequence accuracy in atomic models.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
PhenixCraft is a fully automated pipeline that inserts AlphaFold predictions into Phenix’s map-segmentation step to overcome noise and artifacts, thereby producing atomic models with superior TM-scores and sequence accuracy compared with standard Phenix workflows.
What carries the argument
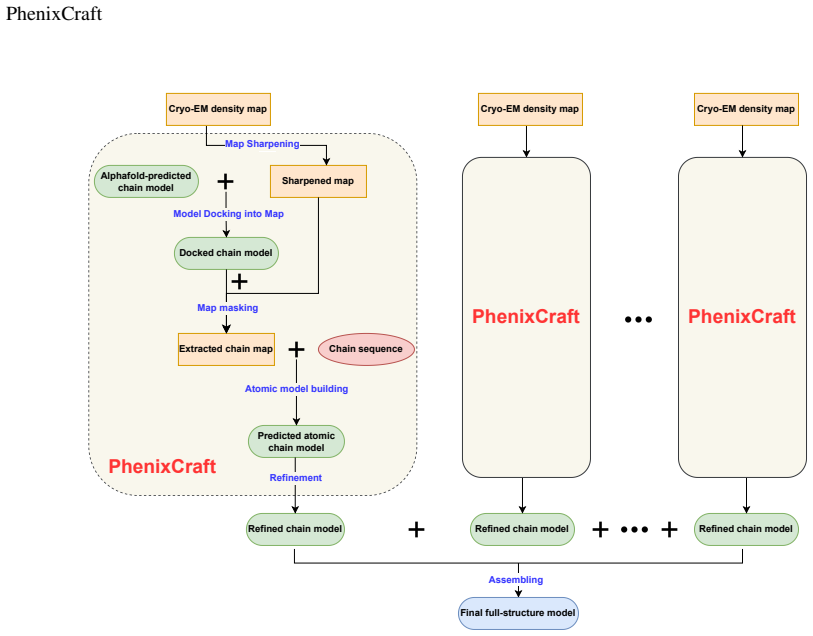
PhenixCraft pipeline, which augments Phenix segmentation by feeding AlphaFold-predicted structures as additional constraints to assign density regions to specific chains or residues.
If this is right
- Atomic models can be completed with less manual intervention when maps contain moderate noise or missing density.
- Sequence accuracy improves because AlphaFold guidance helps assign the correct residues to observed density blobs.
- The pipeline remains fully automated, allowing high-throughput processing of multiple maps without user-guided segmentation.
- Performance gains are demonstrated on the tested cases through direct comparison of TM-scores and sequence matches.
Where Pith is reading between the lines
- The same segmentation-aid idea could be ported to other modeling packages that currently rely on density-thresholding alone.
- If the method tolerates moderate AlphaFold errors, it may still work on lower-resolution maps where experimental density is ambiguous.
- A useful next test would be to quantify how much the gain shrinks when AlphaFold predictions come from more distant homologs rather than close relatives.
Load-bearing premise
AlphaFold predictions are sufficiently accurate that their use in segmentation does not introduce new placement errors or biases into the final atomic model.
What would settle it
A side-by-side test on a benchmark set of cryo-EM maps with deposited ground-truth models, measuring whether PhenixCraft models show statistically higher TM-scores and sequence identity than models built with unmodified Phenix on the same maps.
Figures
read the original abstract
We introduce PhenixCraft, a fully automated pipeline for building atomic models from cryo-EM density maps. By integrating AlphaFold predictions, we enhance the map-segmentation step in Phenix during model building, addressing challenges posed by noise and artifacts that traditionally hinder this step. Our results demonstrate PhenixCraft's superior performance in TM-scores and sequence accuracy, significantly improving upon the limitations and inefficiencies of traditional model building using Phenix.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces PhenixCraft, a fully automated pipeline for building atomic models from cryo-EM density maps. It integrates AlphaFold predictions to enhance the map-segmentation step in Phenix, claiming to address noise and artifacts and demonstrating superior TM-scores and sequence accuracy over traditional Phenix model building.
Significance. If the performance improvements hold under rigorous validation, this work has the potential to enhance the efficiency and accuracy of atomic model building in cryo-EM structural biology by combining deep learning-based predictions with established software tools like Phenix.
major comments (3)
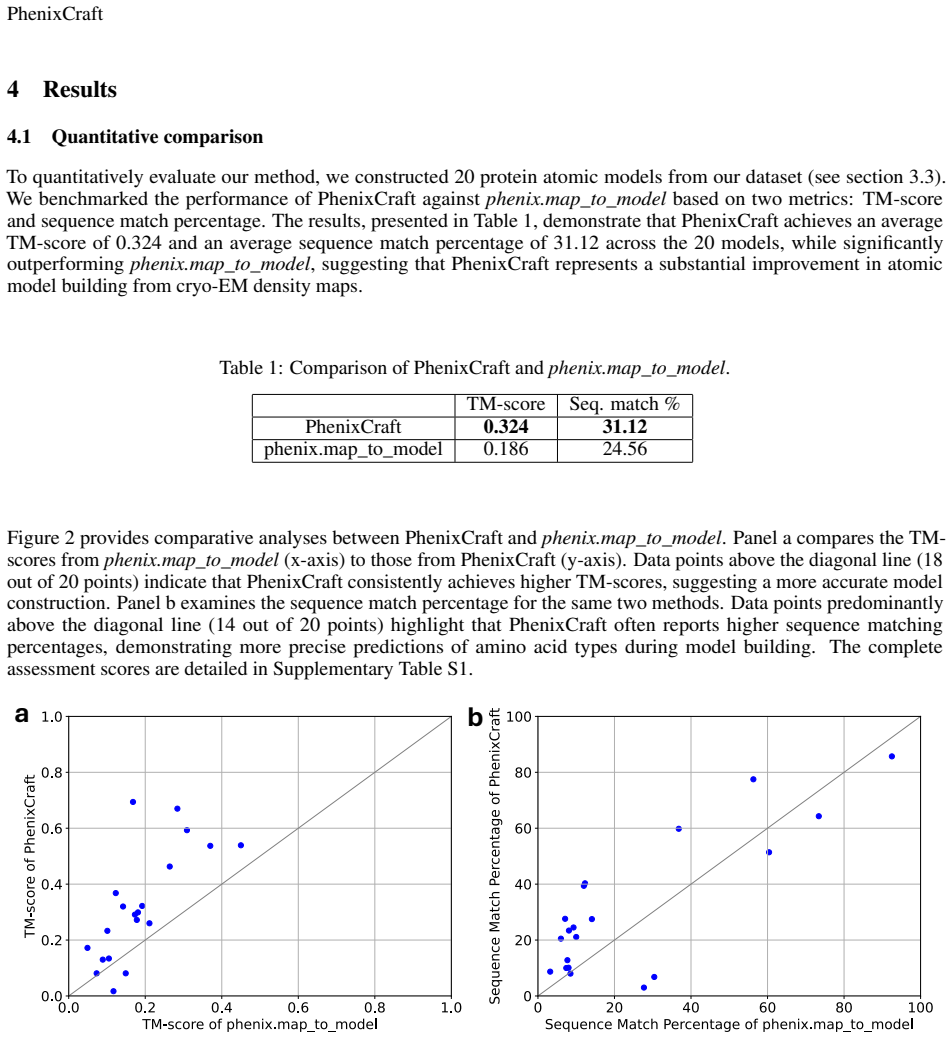
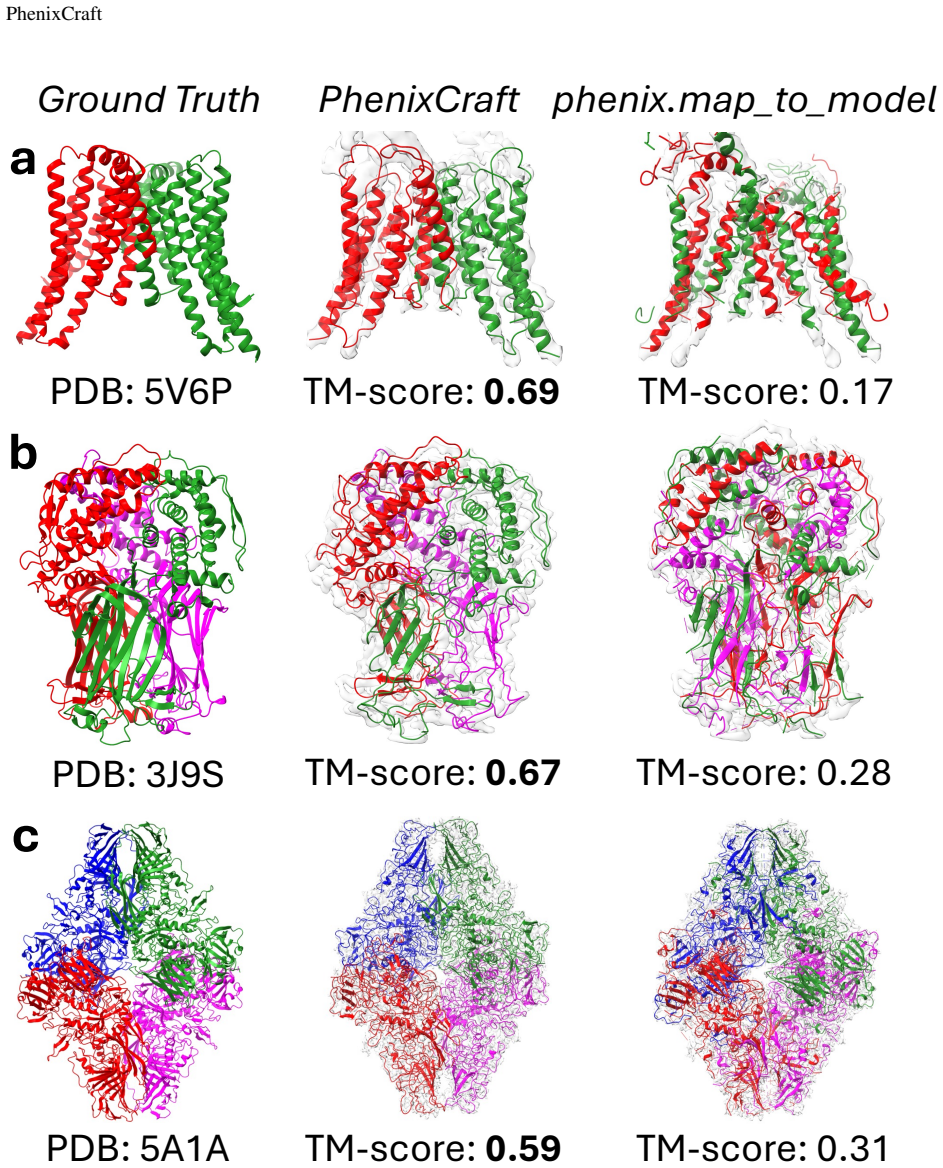
- Abstract: The abstract asserts superior TM-scores and sequence accuracy but provides no quantitative values, test datasets, error bars, baseline comparisons, or statistical tests, making the central performance claim impossible to evaluate from the given information.
- Methods section: The integration of AlphaFold predictions into the Phenix segmentation step is not described in sufficient detail, including the exact mechanism of incorporation and any safeguards against propagating inaccuracies from AlphaFold models (e.g., in low-confidence loop regions or novel folds).
- Results section: No per-region breakdown or control experiments are presented to show that AlphaFold integration improves segmentation without introducing new biases, which is critical given that the segmentation step is identified as the bottleneck.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and constructive comments on our manuscript. We have carefully considered each point and provide detailed responses below, along with plans for revisions to address the concerns raised.
read point-by-point responses
-
Referee: Abstract: The abstract asserts superior TM-scores and sequence accuracy but provides no quantitative values, test datasets, error bars, baseline comparisons, or statistical tests, making the central performance claim impossible to evaluate from the given information.
Authors: We agree with this observation. The abstract in the current version is indeed concise and lacks specific quantitative details. In the revised manuscript, we will update the abstract to include key quantitative results, such as the TM-score improvements and sequence accuracy metrics with error bars, the test datasets used, baseline comparisons to standard Phenix, and mention of the statistical tests applied. This will make the performance claims evaluable from the abstract. revision: yes
-
Referee: Methods section: The integration of AlphaFold predictions into the Phenix segmentation step is not described in sufficient detail, including the exact mechanism of incorporation and any safeguards against propagating inaccuracies from AlphaFold models (e.g., in low-confidence loop regions or novel folds).
Authors: We will revise the Methods section to provide sufficient detail on the integration of AlphaFold predictions into the Phenix segmentation step. This will include the exact mechanism of incorporation as well as safeguards against propagating inaccuracies from AlphaFold models, particularly in low-confidence loop regions or for novel folds. revision: yes
-
Referee: Results section: No per-region breakdown or control experiments are presented to show that AlphaFold integration improves segmentation without introducing new biases, which is critical given that the segmentation step is identified as the bottleneck.
Authors: We agree that additional analyses are needed. In the revised manuscript, we will present per-region breakdowns in the Results section and include control experiments to confirm that the AlphaFold integration improves segmentation without introducing new biases. This is particularly important as the segmentation step is the identified bottleneck. revision: yes
Circularity Check
No circularity: empirical method comparison with external inputs
full rationale
The paper introduces PhenixCraft as a pipeline that feeds AlphaFold predictions into Phenix's map-segmentation step and reports higher TM-scores plus sequence accuracy versus baseline Phenix on test maps. No equations, fitted parameters, uniqueness theorems, or derivation chain exist. AlphaFold is treated as an independent black-box input whose outputs are used as additional restraints; performance numbers are measured against external ground-truth models rather than being forced by the method's own definitions or self-citations. This is a standard applied-methods paper whose central claim is falsifiable by independent benchmarks and does not reduce to its inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption AlphaFold structure predictions are accurate enough to improve density map segmentation in Phenix
invented entities (1)
-
PhenixCraft pipeline
no independent evidence
Reference graph
Works this paper leans on
-
[1]
The resolution revolution.Science, 343(6178):1443–1444, 2014
Werner Kühlbrandt. The resolution revolution.Science, 343(6178):1443–1444, 2014
2014
-
[2]
Overview and future of single particle electron cryomicroscopy.Archives of biochemistry and biophysics, 581:19–24, 2015
Richard Henderson. Overview and future of single particle electron cryomicroscopy.Archives of biochemistry and biophysics, 581:19–24, 2015
2015
-
[3]
Cryo-em: the resolution revolution and drug discovery.SLAS Discovery: Advancing the Science of Drug Discovery, 26(1):17–31, 2021
Taiana Maia de Oliveira, Lotte van Beek, Fiona Shilliday, Judit É Debreczeni, and Chris Phillips. Cryo-em: the resolution revolution and drug discovery.SLAS Discovery: Advancing the Science of Drug Discovery, 26(1):17–31, 2021
2021
-
[4]
Chenwei Zhang, Anne Condon, and Khanh Dao Duc. A comprehensive survey and benchmark of deep learning- based methods for atomic model building from cryo-electron microscopy density maps.Briefings in Bioinformatics, 26(4):bbaf322, 2025
2025
-
[5]
Novel artificial intelligence-based approaches for ab initio structure determination and atomic model building for cryo-electron microscopy.Micromachines, 14(9):1674, 2023
Megan C DiIorio and Arkadiusz W Kulczyk. Novel artificial intelligence-based approaches for ab initio structure determination and atomic model building for cryo-electron microscopy.Micromachines, 14(9):1674, 2023
2023
-
[6]
Alignment of partially overlapping cryo-em maps using unbalanced gromov-wasserstein divergence.PRX Life, 3(2):023003, 2025
Aryan Tajmir Riahi, Chenwei Zhang, Anne Condon, James Chen, and Khanh Dao Duc. Alignment of partially overlapping cryo-em maps using unbalanced gromov-wasserstein divergence.PRX Life, 3(2):023003, 2025
2025
-
[7]
A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps.Nature methods, 15(11):905–908, 2018
Thomas C Terwilliger, Paul D Adams, Pavel V Afonine, and Oleg V Sobolev. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps.Nature methods, 15(11):905–908, 2018
2018
-
[8]
A graph neural network approach to automated model building in cryo-em maps
Kiarash Jamali, Dari Kimanius, and Sjors HW Scheres. A graph neural network approach to automated model building in cryo-em maps. InThe Eleventh International Conference on Learning Representations, 2022
2022
-
[9]
Jonas Pfab, Nhut Minh Phan, and Dong Si. Deeptracer for fast de novo cryo-em protein structure modeling and special studies on cov-related complexes.Proceedings of the National Academy of Sciences, 118(2):e2017525118, 2021
2021
-
[10]
Modeling symmetric macromolecular structures in rosetta3.PloS one, 6(6):e20450, 2011
Frank DiMaio, Andrew Leaver-Fay, Phil Bradley, David Baker, and Ingemar André. Modeling symmetric macromolecular structures in rosetta3.PloS one, 6(6):e20450, 2011
2011
-
[11]
Highly accurate protein structure prediction with alphafold.Nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.Nature, 596(7873):583–589, 2021
2021
-
[12]
Auto-sharpening cryo-em or crystallographic maps with auto_sharpen
Tom Terwilliger et al. Auto-sharpening cryo-em or crystallographic maps with auto_sharpen. https: //phenix-online.org/documentation/reference/auto_sharpen.html. Accessed: 2024
2024
-
[13]
Automated map sharpening by maximization of detail and connectivity.Acta Crystallographica Section D: Structural Biology, 74(6):545–559, 2018
Thomas C Terwilliger, Oleg V Sobolev, Pavel V Afonine, and Paul D Adams. Automated map sharpening by maximization of detail and connectivity.Acta Crystallographica Section D: Structural Biology, 74(6):545–559, 2018
2018
-
[14]
Improvement of cryo-em maps by simultaneous local and non-local deep learning.Nature communications, 14(1):3217, 2023
Jiahua He, Tao Li, and Sheng-You Huang. Improvement of cryo-em maps by simultaneous local and non-local deep learning.Nature communications, 14(1):3217, 2023
2023
-
[15]
Chenwei Zhang and Khanh Dao Duc. Cryosamu: Enhancing 3d cryo-em density maps of protein structures at intermediate resolution with structure-aware multimodal u-nets.arXiv preprint arXiv:2503.20291, 2025
-
[16]
Deepemhancer: a deep learning solution for cryo-em volume post-processing.Communications biology, 4(1):874, 2021
Ruben Sanchez-Garcia, Josue Gomez-Blanco, Ana Cuervo, Jose Maria Carazo, Carlos Oscar S Sorzano, and Javier Vargas. Deepemhancer: a deep learning solution for cryo-em volume post-processing.Communications biology, 4(1):874, 2021
2021
-
[17]
Struc2mapgan: improving synthetic cryogenic electron microscopy density maps with generative adversarial networks.Bioinformatics Advances, 5(1):vbaf179, 2025
Chenwei Zhang, Anne Condon, and Khanh Dao Duc. Struc2mapgan: improving synthetic cryogenic electron microscopy density maps with generative adversarial networks.Bioinformatics Advances, 5(1):vbaf179, 2025
2025
-
[18]
T. C. Terwilliger. Computational crystallography newsletter.Comput. Crystallogr. Newsl., 9:51–57, 2018
2018
-
[19]
phenix.map_box: extract box with model and map around selected atoms
Tom Terwilliger et al. phenix.map_box: extract box with model and map around selected atoms. https: //phenix-online.org/documentation/reference/map_box.html. Accessed: 2024
2024
-
[20]
Emdatabank unified data resource for 3dem
Catherine L Lawson, Ardan Patwardhan, Matthew L Baker, Corey Hryc, Eduardo Sanz Garcia, Brian P Hudson, Ingvar Lagerstedt, Steven J Ludtke, Grigore Pintilie, Raul Sala, et al. Emdatabank unified data resource for 3dem. Nucleic acids research, 44(D1):D396–D403, 2016
2016
-
[21]
The protein data bank.Acta Crystallographica Section D: Biological Crystallography, 58(6):899–907, 2002
Helen M Berman, Tammy Battistuz, Talapady N Bhat, Wolfgang F Bluhm, Philip E Bourne, Kyle Burkhardt, Zukang Feng, Gary L Gilliland, Lisa Iype, Shri Jain, et al. The protein data bank.Acta Crystallographica Section D: Biological Crystallography, 58(6):899–907, 2002. 7 PhenixCraft
2002
-
[22]
Terwilliger
Thomas C. Terwilliger. Phenix.map to model: A fully automatic method yielding initial models from high- resolution electron cryo-microscopy maps. https://phenix-online.org/phenix_data/terwilliger/ map_to_model_2018/, 2018
2018
-
[23]
Colabfold: making protein folding accessible to all.Nature methods, 19(6):679–682, 2022
Milot Mirdita, Konstantin Schütze, Yoshitaka Moriwaki, Lim Heo, Sergey Ovchinnikov, and Martin Steinegger. Colabfold: making protein folding accessible to all.Nature methods, 19(6):679–682, 2022
2022
-
[24]
Ucsf chimerax: Structure visualization for researchers, educators, and developers
Eric F Pettersen, Thomas D Goddard, Conrad C Huang, Elaine C Meng, Gregory S Couch, Tristan I Croll, John H Morris, and Thomas E Ferrin. Ucsf chimerax: Structure visualization for researchers, educators, and developers. Protein Science, 30(1):70–82, 2021
2021
-
[25]
Scoring function for automated assessment of protein structure template quality
Yang Zhang and Jeffrey Skolnick. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics, 57(4):702–710, 2004
2004
-
[26]
Srayanta Mukherjee and Yang Zhang. Mm-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming.Nucleic acids research, 37(11):e83–e83, 2009. 8 PhenixCraft Supplementary Material EMDB: 8782 PDB: 5W81TM-score: 0.30 a b c TM-score: 0.18 Figure S1: In (a), the experimental density map is colored blue....
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.