Recognition: no theorem link
OmicsLM: A Multimodal Large Language Model for Multi-Sample Omics Reasoning
Pith reviewed 2026-05-11 01:10 UTC · model grok-4.3
The pith
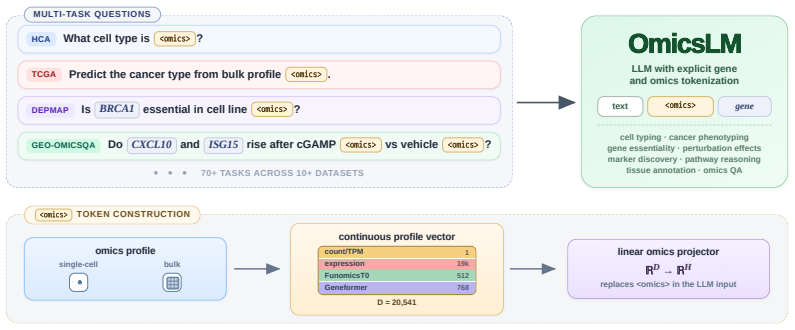
OmicsLM integrates quantitative transcriptomic profiles directly into an LLM context to handle both profile predictions and language-guided reasoning over multiple samples.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
OmicsLM represents each transcriptomic profile as a compact continuous representation within the LLM context. This interface preserves quantitative expression signal while allowing natural-language instructions, explicit gene mentions, and multiple interleaved biological samples to be processed together in one model context. Trained on more than 5.5 million instruction-following examples spanning over 70 task types, OmicsLM performs comparably to specialized omics models on profile-level tasks and outperforms both omics-specialized models and general LLMs on language-guided biological reasoning over expression data.
What carries the argument
The continuous embedding of each transcriptomic profile as a vector inside the LLM context, which lets quantitative data interleave with language instructions and multiple samples.
If this is right
- The model can directly consume expression profiles for cell type annotation, perturbation prediction, and clinical tasks without separate preprocessing steps.
- Language instructions can reference specific genes and compare multiple real expression samples within a single forward pass.
- A new benchmark called GEO-OmicsQA measures language-guided multi-sample reasoning on actual Gene Expression Omnibus studies.
- One unified model can now cover both quantitative profile prediction and open-ended biological question answering.
Where Pith is reading between the lines
- The same continuous-embedding approach could be tested on other high-dimensional biological measurements such as proteomics or metabolomics.
- Biologists might use conversational queries to explore large public datasets without writing code or running separate analysis pipelines.
- Scaling the training mixture to include spatial or single-cell data could extend the same interface to finer-grained biological questions.
Load-bearing premise
Representing each transcriptomic profile as a compact continuous representation preserves enough of the original quantitative expression values to support accurate multi-sample reasoning guided by natural language.
What would settle it
Replacing the continuous profile embeddings with random vectors or text-only summaries and observing that performance on the GEO-OmicsQA benchmark drops to match general LLMs without data access would falsify the claim that the representation carries sufficient signal.
Figures
read the original abstract
Interpreting transcriptomic data is one of the most common analytical tasks in modern biology. Yet most current models either consume expression profiles without producing natural-language biological explanations, or reason in language without direct access to quantitative omics measurements. We introduce OmicsLM, a multimodal LLM that connects quantitative omics profiles with natural-language biological tasks. OmicsLM represents each transcriptomic profile as a compact continuous representation within the LLM context. This interface preserves quantitative expression signal while allowing natural-language instructions, explicit gene mentions, and multiple interleaved biological samples to be processed together in one model context. We train OmicsLM on more than 5.5 million instruction-following examples spanning over 70 task types, combining continuous transcriptomic inputs, experimental data rendered through diverse language templates, and free-text biological knowledge and question-answering data. This mixture covers cell type annotation, perturbation prediction, clinical prediction, pathway reasoning, and open-ended biological question answering. Existing benchmarks evaluate either profile-level prediction or text-only biological QA, leaving language-guided, multi-sample reasoning over real expression profiles unmeasured. To close this gap, we introduce GEO-OmicsQA, a benchmark for multi-sample biological question answering built from real Gene Expression Omnibus (GEO) studies. We demonstrate that OmicsLM can use expression profiles directly and perform comparably to specialized omics models on profile-level tasks, while outperforming both omics-specialized models and general LLMs on language-guided biological reasoning over expression data.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces OmicsLM, a multimodal LLM that processes transcriptomic profiles as compact continuous representations alongside natural language instructions for various biological reasoning tasks. It describes training on a large mixture of instruction data including omics profiles, language templates, and free-text knowledge, and proposes the GEO-OmicsQA benchmark for evaluating multi-sample, language-guided reasoning over real GEO expression data. The central claim is that OmicsLM achieves performance comparable to specialized omics models on profile-level tasks and superior performance on language-guided biological reasoning tasks.
Significance. If substantiated, this work could meaningfully advance integrative biology by enabling direct multimodal reasoning over quantitative expression data with natural-language outputs. The scale of the training mixture (>5.5M examples across 70 tasks) and the introduction of a dedicated benchmark for multi-sample GEO-based QA are constructive contributions that address a genuine evaluation gap. The approach of interleaving continuous profile embeddings with explicit gene mentions and language templates is a reasonable architectural direction for the claimed use cases.
major comments (2)
- [Abstract] Abstract: The abstract asserts that OmicsLM 'perform[s] comparably to specialized omics models on profile-level tasks' and 'outperform[s] both omics-specialized models and general LLMs on language-guided biological reasoning over expression data,' yet supplies no quantitative metrics, baseline details, statistical tests, error bars, or ablation results. Without these, the central claim of effective multimodal integration cannot be evaluated and the reported outperformance on GEO-OmicsQA remains unverifiable.
- [Abstract] Abstract (representation paragraph): The statement that each transcriptomic profile is mapped to a 'compact continuous representation' that 'preserves quantitative expression signal' is load-bearing for the multi-sample reasoning claim, but the manuscript provides no embedding dimension, encoder architecture, reconstruction loss, or ablation that isolates the continuous channel from the language templates also present in training. This omission directly engages the concern that high-dimensional counts may collapse into an insufficient summary, undermining the assertion of true quantitative multimodal reasoning.
Simulated Author's Rebuttal
We thank the referee for the thoughtful review and constructive suggestions. We have revised the abstract to address the concerns about missing quantitative details and technical specifications. Our point-by-point responses are below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The abstract asserts that OmicsLM 'perform[s] comparably to specialized omics models on profile-level tasks' and 'outperform[s] both omics-specialized models and general LLMs on language-guided biological reasoning over expression data,' yet supplies no quantitative metrics, baseline details, statistical tests, error bars, or ablation results. Without these, the central claim of effective multimodal integration cannot be evaluated and the reported outperformance on GEO-OmicsQA remains unverifiable.
Authors: We agree that the abstract would be strengthened by including specific quantitative evidence. In the revised version of the manuscript, we have updated the abstract to report key performance metrics from our experiments, including the specific scores on profile-level tasks where OmicsLM matches specialized models, and the percentage improvements on the GEO-OmicsQA benchmark over both omics-specialized and general LLMs. We also reference the statistical tests and ablations detailed in the main text and supplementary materials. This makes the claims more verifiable while keeping the abstract concise. revision: yes
-
Referee: [Abstract] Abstract (representation paragraph): The statement that each transcriptomic profile is mapped to a 'compact continuous representation' that 'preserves quantitative expression signal' is load-bearing for the multi-sample reasoning claim, but the manuscript provides no embedding dimension, encoder architecture, reconstruction loss, or ablation that isolates the continuous channel from the language templates also present in training. This omission directly engages the concern that high-dimensional counts may collapse into an insufficient summary, undermining the assertion of true quantitative multimodal reasoning.
Authors: We acknowledge this point and have revised the abstract to include a concise description of the representation. The full details of the encoder architecture, embedding dimension, training with reconstruction loss, and the ablation study isolating the continuous embeddings (which demonstrates their contribution to preserving quantitative signal beyond language templates) are provided in the Methods section and Supplementary Information. We believe this addresses the concern about potential collapse of information. revision: yes
Circularity Check
No significant circularity; training mixture and new benchmark are independent of claimed results
full rationale
The paper describes training OmicsLM on a stated mixture of >5.5M instruction examples (continuous profiles + language templates + free-text knowledge) and evaluates on the newly introduced GEO-OmicsQA benchmark built from real GEO studies. No load-bearing step reduces by construction to its own inputs: the continuous embedding is an architectural choice whose fidelity is tested rather than presupposed, the benchmark is external to training data, and performance claims rest on standard held-out evaluation rather than self-referential fitting or self-citation chains. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- omics profile embedding dimension
axioms (1)
- domain assumption Continuous embeddings can preserve sufficient quantitative signal from expression profiles for downstream reasoning tasks
invented entities (1)
-
GEO-OmicsQA benchmark
no independent evidence
Reference graph
Works this paper leans on
-
[1]
doi: 10.1038/75556. Jinze Bai, Shuai Bai, Shusheng Yang, Shijie Wang, Sinan Tan, Peng Wang, Junyang Lin, Chang Zhou, and Jingren Zhou. Qwen-VL: A versatile vision-language model for understanding, localization, text reading, and beyond,
-
[2]
Qwen-VL: A Versatile Vision-Language Model for Understanding, Localization, Text Reading, and Beyond
URLhttps://arxiv.org/abs/2308.12966. Shuai Bai, Yuxuan Cai, Ruizhe Chen, Keqin Chen, Xiong-Hui Chen, Zesen Cheng, Lianghao Deng, Wei Ding, Rongyao Fang, Chang Gao, et al. Qwen3-VL technical report,
work page internal anchor Pith review Pith/arXiv arXiv
-
[3]
URL https://arxiv.org/abs/2511.21631. Amos Bairoch. The Cellosaurus, a cell-line knowledge resource.Journal of Biomolecular Techniques, 29(2):25–38,
work page internal anchor Pith review Pith/arXiv arXiv
-
[4]
doi: 10.7171/jbt.18-2902-002. Tom B Brown, Benjamin Mann, Nick Ryder, Melanie Subbiah, Jared D Kaplan, Prafulla Dhariwal, Arvind Neelakantan, Pranav Shyam, Girish Sastry, Amanda Askell, et al. Language models are few-shot learners. InAdvances in Neural Information Processing Systems, volume 33, pages 1877–1901,
-
[5]
LTS data release 2025-01-30
URL https:// chanzuckerberg.github.io/cellxgene-census/. LTS data release 2025-01-30. H. Chen, M. S. Venkatesh, J. Gómez Ortega, S. V . Mahesh, T. Nandi, R. Madduri, K. Pelka, and C. V . Theodoris. Scaling and quantization of large-scale foundation model enables resource- efficient predictions in network biology.Nature Computational Science,
2025
-
[6]
URL https://www.biorxiv.org/content/10.64898/2026.01.12.699186v1
doi: 10.64898/2026.01.12.699186. URL https://www.biorxiv.org/content/10.64898/2026.01.12.699186v1. Emily Clough, Tanya Barrett, Stephen E Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F Kim, Maxim Tomashevsky, Kimberly A Marshall, Katherine H Phillippy, Patti M Sherman, et al. NCBI GEO: archive for gene expression and epigenomics data sets: 23-year up...
-
[7]
Haotian Cui, Chloe Wang, Hanwen Maan, Kuan Pang, Feng Luo, and Bo Wang
doi: 10.1093/nar/gkad965. Haotian Cui, Chloe Wang, Hanwen Maan, Kuan Pang, Feng Luo, and Bo Wang. scgpt: toward building a foundation model for single-cell multi-omics using generative AI.Nature Methods, 21 (8):1470–1480,
-
[8]
doi: 10.1038/s41592-024-02201-0. Tri Dao. FlashAttention-2: Faster attention with better parallelism and work partitioning. In International Conference on Learning Representations,
-
[9]
URL https://doi.org/10.25452/figshare.plus. 27993248.v1. James Eberwine, Jun-Yong Sul, Tamas Bartfai, and Junhyong Kim. The promise of single-cell sequencing.Nature Methods, 11(1):25–27,
-
[10]
doi: 10.1038/nmeth.2769. Yin Fang, Qiao Jin, Guangzhi Xiong, Bowen Jin, Xianrui Zhong, Siru Ouyang, Aidong Zhang, Jiawei Han, and Zhiyong Lu. Cell-o1: Training LLMs to solve single-cell reasoning puzzles with reinforcement learning,
-
[11]
URLhttps://arxiv.org/abs/2506.02911. 10 Gene Ontology Consortium. The gene ontology knowledgebase in 2026.Nucleic Acids Research, 2025a. doi: 10.1093/nar/gkaf1292. gkaf1292. Gene Ontology Consortium. Gene ontology knowledgebase, 2025b. URL https://doi.org/10. 5281/zenodo.15611813. Google DeepMind. Gemini 3.1 Flash-Lite model card, March 2026a. URL https:/...
-
[12]
doi: 10.1126/science.aaz1776. M. Hao, J. Gong, X. Zeng, W. Liu, J. Liu, T. Chen, et al. Large-scale foundation model on single-cell transcriptomics.Nature Methods, 21:1481–1491,
-
[13]
Leon Hetzel, Simon Böhm, Niki Kilbertus, Stephan Günnemann, Mohammad Lotfollahi, and Fabian Theis
doi: 10.1038/s41592-024-02305-7. Leon Hetzel, Simon Böhm, Niki Kilbertus, Stephan Günnemann, Mohammad Lotfollahi, and Fabian Theis. Predicting cellular responses to novel drug perturbations at a single-cell resolution. InAdvances in Neural Information Processing Systems, volume 35,
-
[14]
URLhttps://doi.org/10.1101/2025.06.11.659105
doi: 10.1101/ 2025.06.11.659105. URLhttps://doi.org/10.1101/2025.06.11.659105. Preprint. Neel Jain, Ping-yeh Chiang, Yuxin Wen, John Kirchenbauer, Hong-Min Chu, Gowthami Somepalli, Brian R Bartoldson, Bhavya Kailkhura, Avi Schwarzschild, Aniruddha Saha, et al. NEFTune: Noisy embeddings improve instruction finetuning. InInternational Conference on Learning...
-
[15]
Boming Kang, Rui Fan, Meizheng Yi, Chunmei Cui, and Qinghua Cui
doi: 10.1093/nar/gkae909. Boming Kang, Rui Fan, Meizheng Yi, Chunmei Cui, and Qinghua Cui. A large-scale foundation model for bulk transcriptomes.bioRxiv,
-
[16]
URL https: //doi.org/10.1101/2025.06.11.659222
doi: 10.1101/2025.06.11.659222. URL https: //doi.org/10.1101/2025.06.11.659222. Preprint. Alexander Lachmann, Denis Torre, Alexandra B Keenan, Kathleen M Jagodnik, Hoyjin J Lee, Lily Wang, Moshe C Silverstein, and Avi Ma’ayan. Massive mining of publicly available RNA- seq data from human and mouse.Nature Communications, 9(1):1366,
-
[17]
URL https://arxiv.org/abs/ 2412.02915. 11 Jianfang Liu, Tara Lichtenberg, Katherine A Hoadley, Laila M Poisson, Alexander J Lazar, Andrew D Cherniack, Albert J Kovatich, Christopher C Benz, Douglas A Levine, Adrian V Lee, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell, 173(2):400–416.e11,
-
[18]
Yang Liu, Dan Iter, Yichong Xu, Shuohang Wang, Ruochen Xu, and Chenguang Zhu
doi: 10.1016/j.cell.2018.02.052. Yang Liu, Dan Iter, Yichong Xu, Shuohang Wang, Ruochen Xu, and Chenguang Zhu. G-Eval: NLG evaluation using GPT-4 with better human alignment. InProceedings of the 2023 Confer- ence on Empirical Methods in Natural Language Processing, pages 2511–2522. Association for Computational Linguistics, 2023b. doi: 10.18653/v1/2023.e...
-
[19]
Ajay Nadig, Joseph M Replogle, Angela N Pogson, et al
doi: 10.1093/nar/gkae1078. Ajay Nadig, Joseph M Replogle, Angela N Pogson, et al. Transcriptome-wide analysis of differential expression in perturbation atlases.Nature Genetics, 57:1228–1237,
-
[20]
doi: 10.1038/s41588-025-02169-3. OpenAI. GPT-4 technical report.arXiv preprint arXiv:2303.08774,
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1038/s41588-025-02169-3
-
[21]
URL https://arxiv. org/abs/2303.08774. OpenAI. GPT-5.5 system card,
work page internal anchor Pith review Pith/arXiv arXiv
-
[22]
Simultaneous learning from bulk and single-cell expression data with perceiver-based models
Rafał Powalski, Albert Roethel, Joanna Krawczyk, Przemysław Pietrzak, Łukasz Smoli´nski, Maciej Sypetkowski, Paweł Biernat, Dariusz Plewczy´nski, and Tomasz Jetka. Simultaneous learning from bulk and single-cell expression data with perceiver-based models. InICLR 2026 Workshop on Machine Learning for Genomics Explorations (MLGenX),
2026
-
[23]
URLhttps://arxiv.org/abs/2505.09388. Samyam Rajbhandari, Jeff Rasley, Olatunji Ruwase, and Yuxiong He. Zero: memory optimizations toward training trillion parameter models. InProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, SC ’20. IEEE Press,
work page internal anchor Pith review Pith/arXiv arXiv
-
[24]
doi: 10.7554/eLife.27041. Joseph M Replogle, Reuben A Saunders, Angela N Pogson, Jeffrey A Hussmann, Alexander Lenail, Alina Guna, Lauren Mascibroda, Eric J Wagner, Karen Adelman, Gila Lithwick-Yanai, et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq.Cell, 185(14):2559–2575.e28,
-
[25]
doi: 10.1016/j.cell.2022.05.013. Syed Asad Rizvi, Daniel Levine, Aakash Patel, Shiyang Zhang, Eric Wang, Sizhuang He, David Zhang, Cerise Tang, Zhuoyang Lyu, Rayyan Darji, Chang Li, Emily Sun, David Jeong, Lawrence Zhao, Jennifer Kwan, David Braun, Brian Hafler, Jeffrey Ishizuka, Rahul M. Dhodapkar, Hattie 12 Chung, Shekoofeh Azizi, Bryan Perozzi, and Dav...
-
[26]
URL https://www.biorxiv.org/content/10.1101/2025.04.14.648850v2
doi: 10.1101/2025.04.14.648850. URL https://www.biorxiv.org/content/10.1101/2025.04.14.648850v2. Yanay Rosen, Yusuf Roohani, Ayush Agrawal, Leon Samotorcan, Tabula Sapiens Consortium, Stephen R Quake, and Jure Leskovec. Universal cell embeddings: A foundation model for cell biology.bioRxiv,
-
[27]
Universal Cell Embeddings: A Foundation Model for Cell Biology
doi: 10.1101/2023.11.28.568918. URL https://www.biorxiv.org/ content/10.1101/2023.11.28.568918v1. M. Schaefer, P. Peneder, D. Malzl, S. D. Lombardo, M. Peycheva, J. Burton, A. Hakobyan, V . Sharma, T. Krausgruber, C. Sin, J. Menche, E. M. Tomazou, and C. Bock. Multimodal learning enables chat-based exploration of single-cell data.Nature Biotechnology,
-
[28]
doi: 10.1038/s41587-025-02857-9. Damian Szklarczyk, Rebecca Kirsch, Mikaela Koutrouli, Katerina Nastou, Farrokh Mehryary, Radja Hachilif, Annika L Gable, Tao Fang, Nadezhda T Doncheva, Sampo Pyysalo, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest.Nucleic Acid...
-
[29]
Nucleic Acids Research , author =
doi: 10.1093/nar/gkac1000. The Tabula Sapiens Consortium. The tabula sapiens: A multiple-organ, single-cell transcriptomic atlas of humans.Science, 376(6594):eabl4896,
-
[30]
doi: 10.1126/science.abl4896. The UniProt Consortium. UniProt: the universal protein knowledgebase in 2025.Nucleic Acids Research, 53(D1):D609–D617,
-
[31]
doi: 10.1093/nar/gkae1010. Christina V . Theodoris, Ling Xiao, Anant Chopra, Mark D. Chaffin, Zeina R. Al Sayed, Matthew C. Hill, Helene Mantineo, Elizabeth M. Brydon, Zexian Zeng, X. Shirley Liu, and Patrick T. Ellinor. Transfer learning enables predictions in network biology.Nature, 618:616–624,
-
[32]
doi: 10.1038/s41586-023-06139-9. Hugo Touvron, Thibaut Lavril, Gautier Izacard, Xavier Martinet, Marie-Anne Lachaux, Timothée Lacroix, Baptiste Rozière, Naman Goyal, Eric Hambro, Faisal Azhar, et al. LLaMA: Open and efficient foundation language models,
-
[33]
LLaMA: Open and Efficient Foundation Language Models
URLhttps://arxiv.org/abs/2302.13971. Eric Wang, Samuel Schmidgall, Paul F Jaeger, Fan Zhang, Rory Pilgrim, Yossi Matias, Joelle Barral, David Fleet, and Shekoofeh Azizi. TxGemma: Efficient and agentic LLMs for therapeutics,
work page internal anchor Pith review Pith/arXiv arXiv
-
[34]
arXiv preprint arXiv:2504.06196 (2025)
URLhttps://arxiv.org/abs/2504.06196. John N Weinstein, Eric A Collisson, Gordon B Mills, Kenna R Mills Shaw, Brad A Ozenberger, Kyle Ellrott, Ilya Shmulevich, Chris Sander, Joshua M Stuart, et al. The cancer genome atlas pan-cancer analysis project.Nature genetics, 45(10):1113–1120,
-
[35]
Fan Yang, Wenchuan Wang, Fang Wang, Yuan Fang, Duyu Tang, Junzhou Huang, Hui Lu, and Jianhua Yao
URL https://arxiv.org/abs/ 2602.09063. Fan Yang, Wenchuan Wang, Fang Wang, Yuan Fang, Duyu Tang, Junzhou Huang, Hui Lu, and Jianhua Yao. scbert as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data.Nature Machine Intelligence, 4:852–866,
-
[36]
doi: 10.1038/s42256-022-00534-z. Fan Zhang, Tianyu Liu, Zhihong Zhu, Hao Wu, Haixin Wang, Donghao Zhou, Yefeng Zheng, Kun Wang, Xian Wu, and Pheng-Ann Heng. Cellverse: Do large language models really understand cell biology?,
-
[37]
Given this expression profile, identify the cell type / tissue / cell-cycle phase
URLhttps://arxiv.org/abs/2505.07865. Haiteng Zhao, Chang Ma, Fangzhi Xu, Lingpeng Kong, and Zhi-Hong Deng. BioMaze: Bench- marking and enhancing large language models for biological pathway reasoning, 2025a. URL https://arxiv.org/abs/2502.16660. Haiteng Zhao, Chang Ma, Fangzhi Xu, Lingpeng Kong, and Zhi-Hong Deng. BioMaze dataset. https://huggingface.co/d...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.