Recognition: no theorem link
Accelerating Bayesian Phylogenetic Inference via Delayed Acceptance Sequential Monte Carlo with Random Forest Surrogates
Pith reviewed 2026-05-12 04:44 UTC · model grok-4.3
The pith
A random forest surrogate predicts likelihood changes from tree moves to let delayed-acceptance SMC reject poor proposals early while preserving the posterior.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
A random forest trained on topological and branch-length features predicts the sign and rough magnitude of the log-likelihood ratio for eSPR and stNNI moves. This predictor is inserted into a delayed-acceptance kernel inside an SMC sampler, so that only moves passing the cheap surrogate test receive a full likelihood evaluation.
What carries the argument
The random forest surrogate that approximates the likelihood change for proposed phylogenetic tree moves, enabling preliminary rejection in the delayed acceptance step.
If this is right
- The delayed-acceptance SMC recovers posterior distributions statistically indistinguishable from those of standard SMC.
- The number of full likelihood evaluations drops substantially, producing measurable reductions in computational time.
- The method performs consistently on both simulated alignments and empirical sequence data.
- The surrogate can be retrained for any chosen collection of tree-move features.
Where Pith is reading between the lines
- The same surrogate strategy could be paired with other expensive likelihood models outside phylogenetics.
- If surrogate bias stays negligible, the delayed-acceptance template supplies a reusable pattern for accelerating any sampler whose proposals admit cheap side information.
- Extending the feature set to datasets with hundreds of taxa would require checking whether the random forest remains unbiased at larger scales.
Load-bearing premise
The random forest surrogate, trained on topological and branch-length features, accurately predicts the sign and rough magnitude of likelihood change for standard tree moves without introducing systematic bias into the posterior.
What would settle it
Run both DA-SMC and ordinary SMC on identical data sets and compare the resulting posterior distributions over trees; any systematic shift in clade support or branch-length quantiles would show that the surrogate has biased the sampler.
Figures
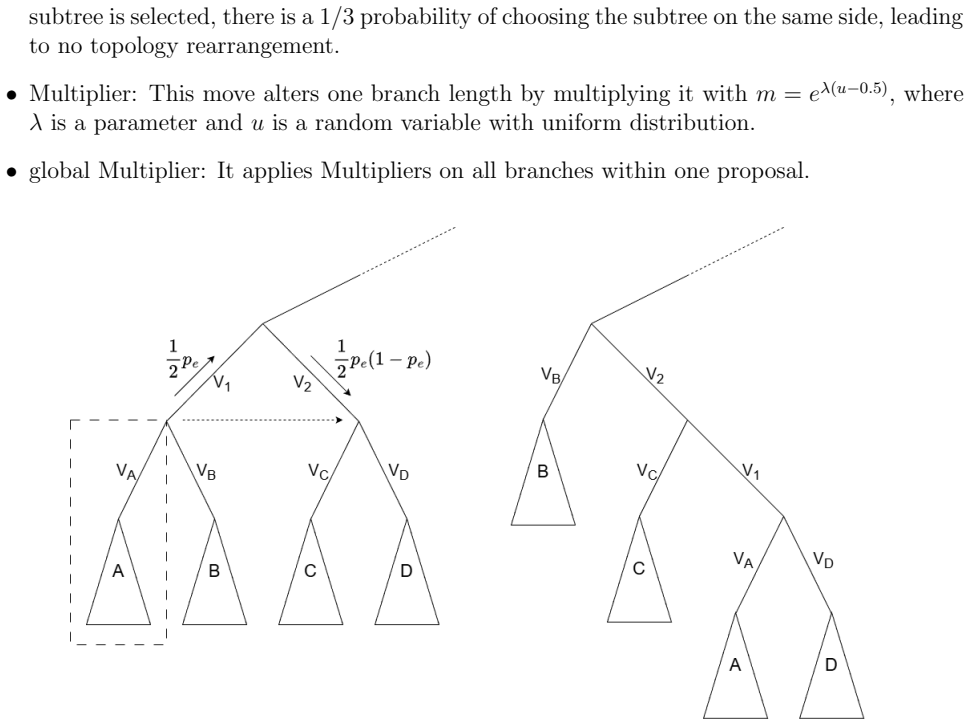
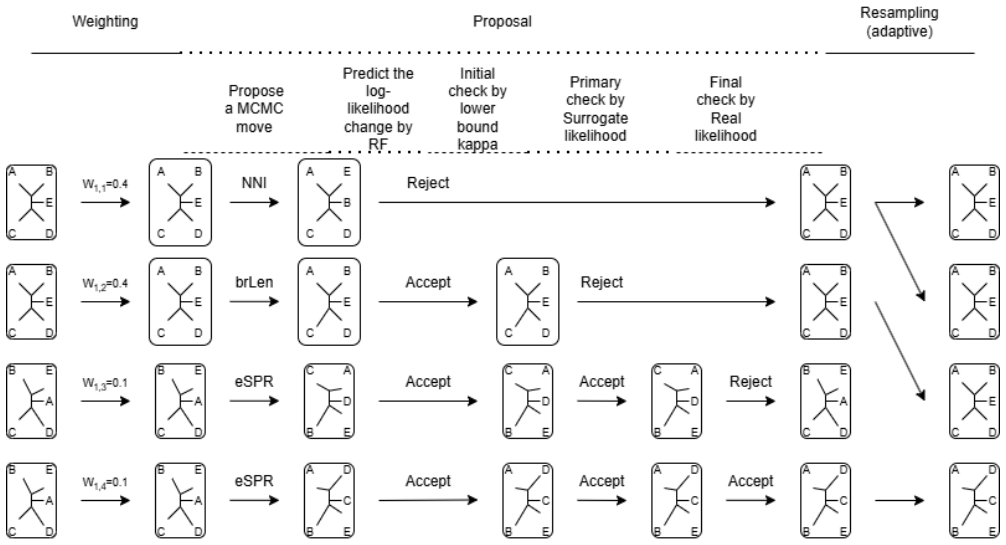
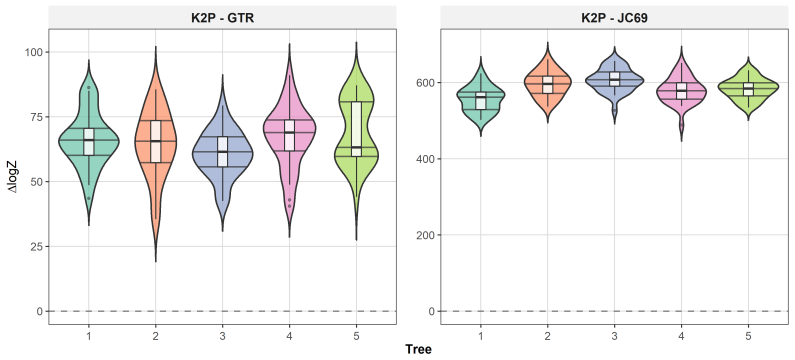
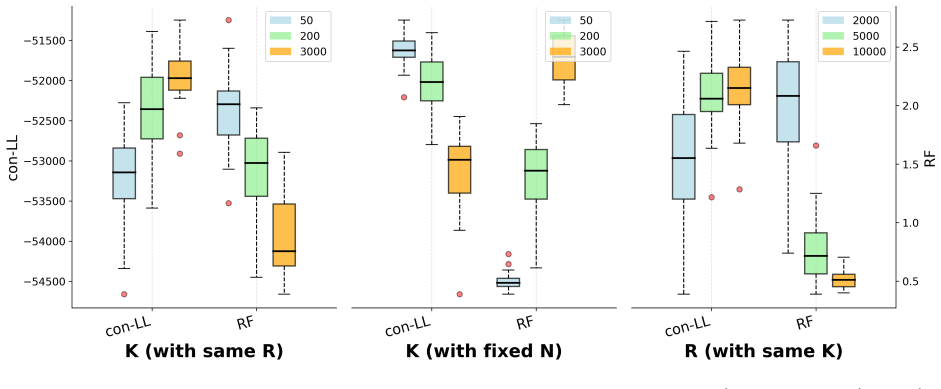
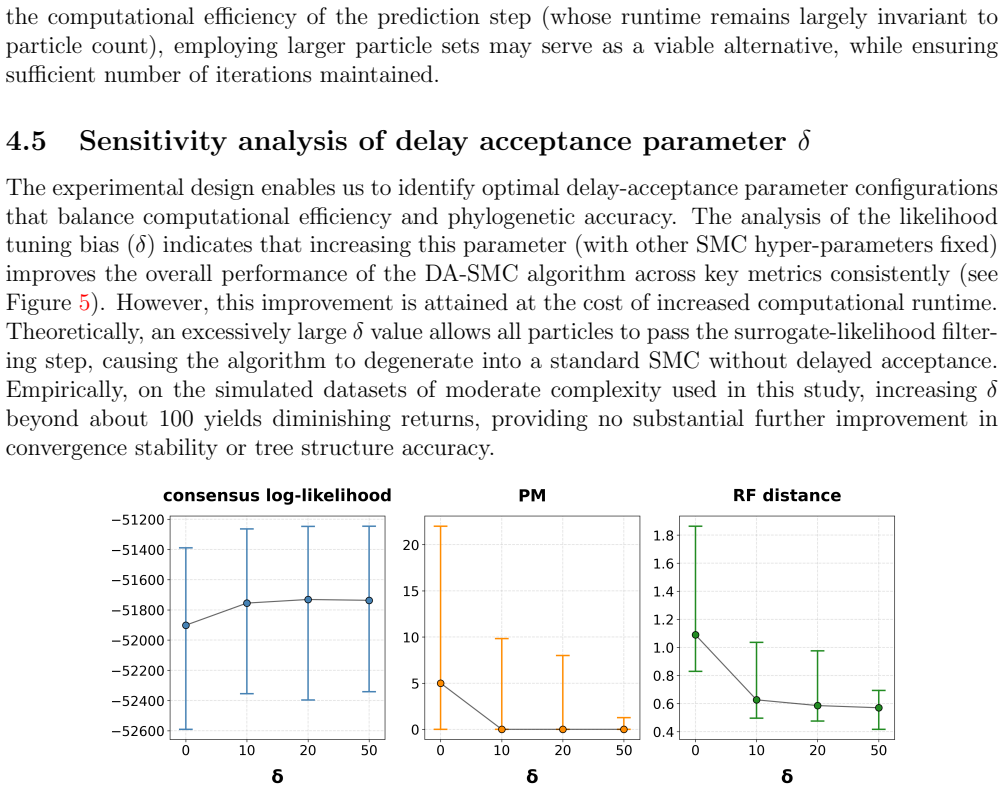

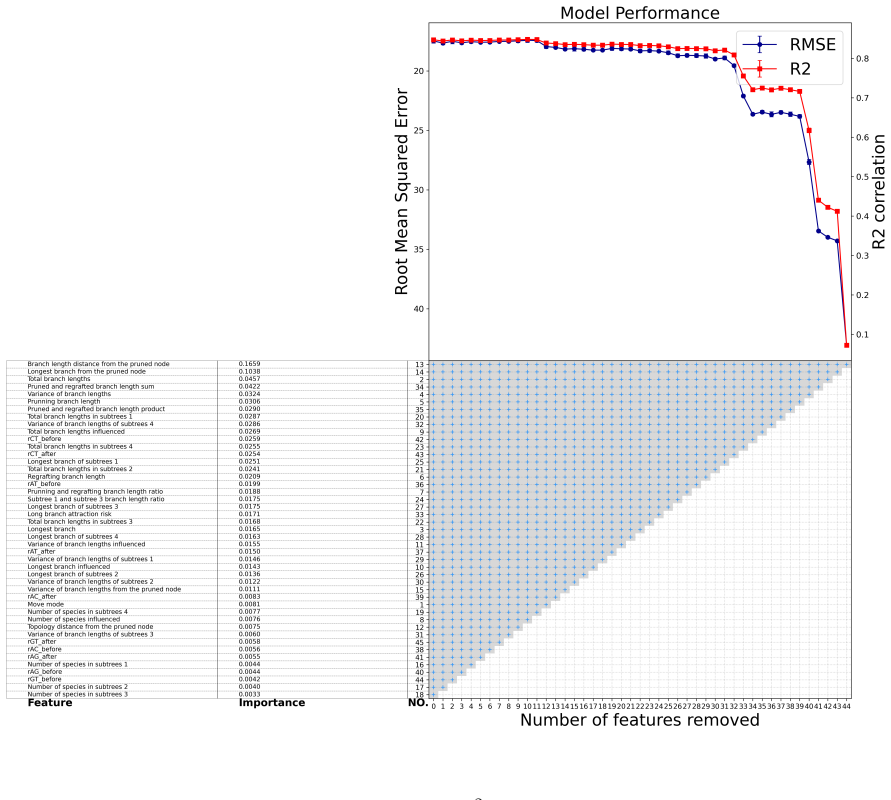
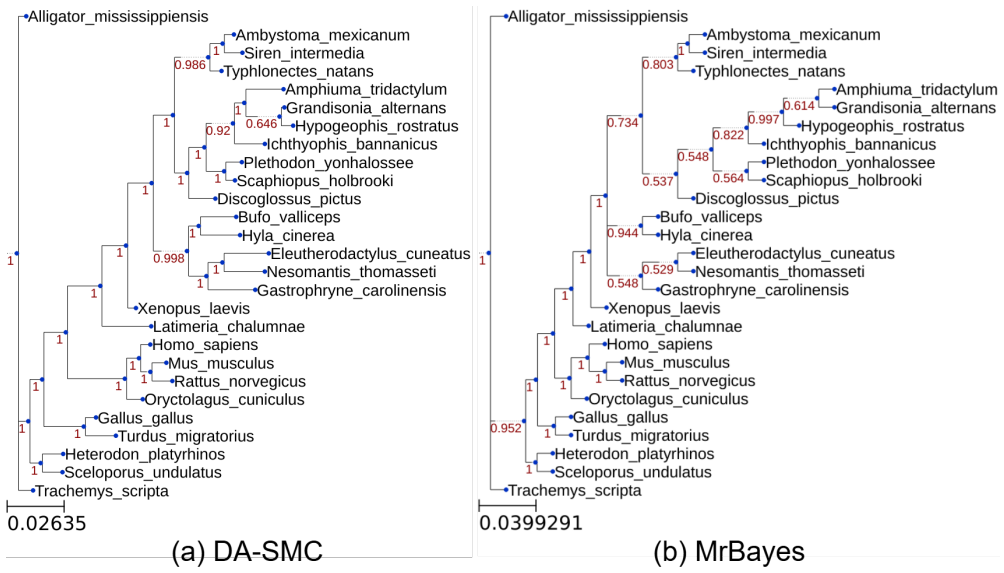
read the original abstract
In Bayesian phylogenetics, our goal is to estimate the posterior distribution over phylogenetic trees. Markov chain Monte Carlo methods are widely used to approximate the phylogenetic posterior distributions. For large-scale sequence data, repeated evaluation of the likelihood function incurs a high computational cost. In this article, we propose a machine-learning algorithm with over 35 topological and branch-length features to predict the changes in the likelihood function caused by tree moves (\eg,~eSPR, stNNI) used in standard MCMC approaches. This algorithm is then used to design a delayed acceptance MCMC kernel, which utilized the predicted surrogate function for preliminary rejection, to accelerate tree space searches. Furthermore, we integrate our proposed MCMC kernel into the sequential Monte Carlo sampler framework. We validate the proposed delayed-acceptance sequential Monte Carlo approach (DA-SMC) on simulation and real data sets. Our delayed acceptance kernel can maintain robust estimation while reduces the number of likelihood evaluations significantly, yielding substantial computational time savings. We develop a Python package that is available at https://github.com/wentYu/DAphyloSMC.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes a delayed-acceptance sequential Monte Carlo (DA-SMC) sampler for Bayesian phylogenetic inference. A random forest surrogate trained on >35 topological and branch-length features predicts the sign and rough magnitude of likelihood changes under standard tree moves (eSPR, stNNI). The surrogate is used only for a first-stage rejection step inside a delayed-acceptance MCMC kernel; the true likelihood is still evaluated in the second stage. This kernel is embedded in an SMC framework to reduce the number of expensive likelihood evaluations while targeting the same posterior. Validation on simulated and real data is reported to show substantial computational savings with maintained estimation quality; an open-source Python package is provided.
Significance. If the surrogate filter does not introduce systematic bias into the effective proposal distribution, the method offers a practical route to accelerate tree-space exploration for large alignments without altering the target posterior. The explicit separation of surrogate and true-likelihood stages, together with the released code, supports reproducibility and further benchmarking.
major comments (2)
- [Results] Results section: the claim that DA-SMC 'maintains robust estimation' is supported only by qualitative statements of reduced evaluations and time savings; no quantitative diagnostics (bias in posterior tree summaries, coverage probabilities, ESS ratios, or posterior-distance metrics relative to standard SMC) are supplied. Without these, it is impossible to verify that surrogate errors do not distort the stationary distribution before the second-stage correction.
- [Methods] Surrogate model description (Methods): the random forest is trained on independent features and used only for preliminary rejection, yet no calibration plots, confusion matrices, or topology/branch-length stratified error rates are reported. If the surrogate systematically under-predicts positive likelihood deltas on large trees or certain topologies, the effective proposal distribution becomes biased even though the final acceptance uses the true likelihood.
minor comments (3)
- [Methods] Notation for the delayed-acceptance kernel (e.g., the two-stage acceptance probability) should be written explicitly with the surrogate and true likelihood distinguished, rather than left implicit.
- [Abstract and Results] The abstract and results would benefit from a short table summarizing wall-clock time, number of likelihood calls, and at least one posterior quality metric for DA-SMC versus baseline SMC on each dataset.
- [Introduction] A few sentences placing the work against existing surrogate-assisted MCMC or SMC methods in phylogenetics would clarify the incremental contribution.
Simulated Author's Rebuttal
We thank the referee for their thoughtful and constructive review. The major comments correctly identify areas where additional quantitative evidence and surrogate diagnostics would strengthen the manuscript. We address each point below and will incorporate the suggested revisions.
read point-by-point responses
-
Referee: [Results] Results section: the claim that DA-SMC 'maintains robust estimation' is supported only by qualitative statements of reduced evaluations and time savings; no quantitative diagnostics (bias in posterior tree summaries, coverage probabilities, ESS ratios, or posterior-distance metrics relative to standard SMC) are supplied. Without these, it is impossible to verify that surrogate errors do not distort the stationary distribution before the second-stage correction.
Authors: We agree that quantitative diagnostics are necessary to rigorously demonstrate that the delayed-acceptance correction preserves the target posterior. Our current validation shows comparable posterior summaries and substantial reductions in likelihood evaluations on both simulated and real data, but we did not report formal bias, coverage, or distance metrics. In the revised manuscript we will add, for the simulation experiments, (i) bias and coverage for key posterior quantities such as clade probabilities, (ii) ESS ratios between DA-SMC and standard SMC, and (iii) posterior-distance metrics (e.g., average Robinson-Foulds distance between independent runs). These additions will appear in the Results section and will be supported by the released code. revision: yes
-
Referee: [Methods] Surrogate model description (Methods): the random forest is trained on independent features and used only for preliminary rejection, yet no calibration plots, confusion matrices, or topology/branch-length stratified error rates are reported. If the surrogate systematically under-predicts positive likelihood deltas on large trees or certain topologies, the effective proposal distribution becomes biased even though the final acceptance uses the true likelihood.
Authors: We acknowledge that explicit surrogate performance diagnostics were not included. Although the two-stage delayed-acceptance construction guarantees that the true likelihood is used for final acceptance and therefore the target posterior remains exact, reporting surrogate accuracy is valuable for assessing efficiency and potential bias in the first-stage filter. In the revision we will add (i) calibration plots of predicted versus true likelihood change, (ii) confusion matrices for sign prediction, and (iii) error rates stratified by tree size and move type (eSPR versus stNNI). These will be placed in the Methods section or a supplementary figure. revision: yes
Circularity Check
No circularity: surrogate accelerates but final posterior uses true likelihood
full rationale
The paper trains a random forest on topological and branch-length features to predict likelihood deltas for eSPR/stNNI moves, then deploys it only for first-stage rejection inside a delayed-acceptance kernel whose second stage always evaluates the exact likelihood. Because the target stationary distribution is recovered from the true-likelihood accept/reject decisions (and the SMC weights are likewise computed with the true likelihood), the fitted surrogate never enters the final posterior by construction. Validation is performed on held-out simulations and real datasets rather than on the training data itself, and no self-citations or uniqueness theorems are invoked to justify the method. The workflow therefore remains independent of its own fitted parameters.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Standard phylogenetic tree moves (eSPR, stNNI) and the likelihood function under common substitution models are well-defined and computable.
- domain assumption A random forest trained on hand-crafted topological and branch-length features can generalize to unseen tree proposals within the same data regime.
Reference graph
Works this paper leans on
-
[1]
Harnessing machine learning to guide phylogenetic-tree search algorithms
Azouri, D., Abadi, S., Mansour, Y., Mayrose, I., and Pupko, T. Harnessing machine learning to guide phylogenetic-tree search algorithms. Nature Communications, 12 0 (1): 0 1983, 2021. doi:10.1038/s41467-021-22073-8. URL https://doi.org/10.1038/s41467-021-22073-8
-
[2]
Bon, J. J., Lee, A., and Drovandi, C. Accelerating sequential monte carlo with surrogate likelihoods. Statistics and Computing, 31 0 (5): 0 62, 2021. doi:10.1007/s11222-021-10036-4. URL https://doi.org/10.1007/s11222-021-10036-4
-
[3]
Bouchard-C\^ot\'e, A., Sankararaman, S., and Jordan, M. I. P hylogenetic inference via sequential M onte C arlo. Systematic Biology, 61: 0 579--593, 2012
work page 2012
-
[4]
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duch \^e ne, S., Fourment, M., Gavryushkina, A., Heled, J., Jones, G., K \"u hnert, D., De Maio, N., et al. BEAST 2.5: An advanced software platform for B ayesian evolutionary analysis. PLoS computational biology, 15 0 (4): 0 e1006650, 2019
work page 2019
- [5]
-
[6]
Using early rejection markov chain monte carlo and gaussian processes to accelerate abc methods
Cao, X., Wang, S., and Zhou, Y. Using early rejection markov chain monte carlo and gaussian processes to accelerate abc methods. Journal of Computational and Graphical Statistics, 34 0 (2): 0 395--408, 2025. doi:10.1080/10618600.2024.2379349. URL https://doi.org/10.1080/10618600.2024.2379349
-
[7]
Chopin, N. Central limit theorem for sequential M onte C arlo methods and its application to B ayesian inference. The Annals of Statistics, 32 0 (6): 0 2385--2411, 2004
work page 2004
-
[8]
Christen, J. A. and Fox, C. Markov chain monte carlo using an approximation. Journal of Computational and Graphical Statistics, 14 0 (4): 0 795--810, 2005. doi:10.1198/106186005X76983. URL https://doi.org/10.1198/106186005X76983
-
[9]
Feynman-Kac Formulae: Genealogical and Interacting Particle Systems with Applications
Del Moral, P. Feynman-Kac Formulae: Genealogical and Interacting Particle Systems with Applications. Springer, New York, 2004
work page 2004
-
[10]
Sequential M onte C arlo samplers
Del Moral, P., Doucet, A., and Jasra, A. Sequential M onte C arlo samplers. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 68 0 (3): 0 411--436, 2006
work page 2006
-
[11]
Dinh, V., Darling, A. E., and Matsen IV, F. A. Online B ayesian phylogenetic inference: theoretical foundations via S equential M onte C arlo. Systematic Biology, 67 0 (3): 0 503--517, 2017
work page 2017
-
[12]
Sequential M onte C arlo methods in practice
Doucet, A., de Freitas, N., and Gordon, N. Sequential M onte C arlo methods in practice . Springer-Verlag, New York, 2001
work page 2001
-
[13]
Drummond, A. and Suchard, M. Bayesian random local clocks, or one rate to rule them all. BMC biology, 8 0 (1): 0 114, 2010
work page 2010
-
[14]
G., Culliford, R., Medina-Aguayo, F., and Wilson, D
Everitt, R. G., Culliford, R., Medina-Aguayo, F., and Wilson, D. J. Sequential B ayesian inference for mixture models and the coalescent using sequential M onte C arlo samplers with transformations. Statistics and Computing, 30 0 (3): 0 663--676, 2020
work page 2020
-
[15]
Felsenstein, J. Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters. Systematic Biology, 22 0 (3): 0 240--249, 1973
work page 1973
-
[16]
Evolutionary trees from DNA sequences: a maximum likelihood approach
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. Journal of molecular evolution, 17 0 (6): 0 368--376, 1981
work page 1981
-
[17]
Foster, P. G. Modeling compositional heterogeneity. Systematic Biology, 53 0 (3): 0 485--495, 06 2004. ISSN 1063-5157. doi:10.1080/10635150490445779. URL https://doi.org/10.1080/10635150490445779
-
[18]
C., Dinh, V., McCoy, C., Matsen, I., Frederick, A., and Darling, A
Fourment, M., Claywell, B. C., Dinh, V., McCoy, C., Matsen, I., Frederick, A., and Darling, A. E. Effective online B ayesian phylogenetics via sequential M onte C arlo with guided proposals. Systematic biology, 2017
work page 2017
-
[19]
Scalable inference on K ingman's coalescent using pair similarity
G\" o r\" u r, D., Boyles, L., and Welling, M. Scalable inference on K ingman's coalescent using pair similarity. Journal of Machine Learning Research, 22: 0 440--448, 2012
work page 2012
-
[20]
G\" o r\" u r, D. and Teh, Y. W. An efficient sequential M onte C arlo algorithm for coalescent clustering. In Advances in Neural Information Processing Systems (NIPS), 2009
work page 2009
-
[21]
Efficient continuous-time M arkov chain estimation
Hajiaghayi, M., Kirkpatrick, B., Wang, L., and Bouchard-C\^ o t\' e , A. Efficient continuous-time M arkov chain estimation. In International Conference on Machine Learning (ICML), volume 31, pages 638--646, 2014
work page 2014
-
[22]
Holder, M. T. Dendropy: a python library for phylogenetic computing. Bioinformatics, 26 0 (12): 0 1569, 2010
work page 2010
-
[23]
Huelsenbeck, J. P. and Ronquist, F. MRBAYES: B ayesian inference of phylogenetic trees. Bioinformatics, 17 0 (8): 0 754--755, 2001
work page 2001
-
[24]
Jukes, T. H., Cantor, C. R., et al. Evolution of protein molecules. Mammalian protein metabolism, 3 0 (21): 0 132, 1969
work page 1969
-
[25]
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of molecular evolution, 16 0 (2): 0 111--120, 1980
work page 1980
-
[26]
P., Larget, B., and Ronquist, F
Lakner, C., van der Mark, P., Huelsenbeck, J. P., Larget, B., and Ronquist, F. Efficiency of M arkov chain M onte C arlo tree proposals in B ayesian phylogenetics. Syst. Biol., 57 0 (1): 0 86--103, 2008
work page 2008
-
[27]
Lemey, P., Rambaut, A., Drummond, A. J., and Suchard, M. A. Bayesian phylogeography finds its roots . PLoS Computational Biology, 5 0 (9): 0 e1000520, 2009. doi:10.1371/journal.pcbi.1000520. URL http://dx.doi.org/10.1371/journal.pcbi.1000520
-
[28]
Adaptive tree proposals for bayesian phylogenetic inference
Meyer, X. Adaptive tree proposals for bayesian phylogenetic inference. Systematic Biology, 70 0 (5): 0 1015--1032, 01 2021. ISSN 1063-5157. doi:10.1093/sysbio/syab004. URL https://doi.org/10.1093/sysbio/syab004
-
[29]
Pedersen, A. G. sumt: a command-line program for computing consensus trees and other phylogenetic tree summaries , November 2023. URL https://github.com/agormp/sumt
work page 2023
-
[30]
Piel, W. H., Chan, L., Dominus, M. J., Ruan, J., Vos, R. A., and Tannen, V. Treebase v. 2: A database of phylogenetic knowledge. In Proceedings of the e-BioSphere 2009, London, UK, 2009. Available at https://treebase.org
work page 2009
-
[31]
Rannala, B. and Yang, Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J. Mol. E, 43: 0 304--311, 1996
work page 1996
-
[32]
Robinson, D. F. and Foulds, L. R. Comparison of weighted labelled trees. In Horadam, A. F. and Wallis, W. D., editors, Combinatorial Mathematics VI, pages 119--126, Berlin, Heidelberg, 1979. Springer Berlin Heidelberg
work page 1979
-
[33]
Robinson, D. and Foulds, L. Comparison of phylogenetic trees. Mathematical Biosciences, 53: 0 131--147, 1981
work page 1981
-
[34]
Rodriguez, F., Oliver, J. L., Marin, A., and Medina, J. R. The general stochastic model of nucleotide substitution. Journal of theoretical biology, 142 0 (4): 0 485--501, 1990
work page 1990
-
[35]
Ronquist, F. and Huelsenbeck, J. P. M r B ayes 3: B ayesian phylogenetic inference under mixed models. Bioinformatics, 19 0 (12): 0 1572--1574, 2003
work page 2003
-
[36]
L., Darling, A., Hohna, S., Larget, B., Liu, L., Suchard, M
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., Larget, B., Liu, L., Suchard, M. A., and Huelsenbeck, J. P. Mr B ayes 3.2: Efficient B ayesian phylogenetic inference and model choice across a large model space. Syst. Biol., 61: 0 539--542, 2012
work page 2012
-
[37]
Smith, R. A., Ionides, E. L., and King, A. A. Infectious disease dynamics inferred from genetic data via sequential M onte C arlo. Molecular biology and evolution, 34 0 (8): 0 2065--2084, 2017
work page 2065
-
[38]
Suchard, M. A. and Redelings, B. D. BA li- P hy: simultaneous B ayesian inference of alignment and phylogeny. Bioinformatics, 22 0 (16): 0 2047--2048, 2006
work page 2047
-
[39]
A., Lemey, P., Baele, G., Ayres, D
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus evolution, 4 0 (1): 0 vey016, 2018
work page 2018
-
[40]
W., Daum\'e III , H., and Roy, D
Teh, Y. W., Daum\'e III , H., and Roy, D. M. B ayesian agglomerative clustering with coalescents. In Advances in Neural Information Processing Systems (NIPS), 2008
work page 2008
-
[41]
Wang, L., Bouchard-C\^ot\'e, A., and Doucet, A. Bayesian phylogenetic inference using a combinatorial sequential M onte C arlo method. Journal of the American Statistical Association, 110 0 (512): 0 1362--1374, 2015. doi:10.1080/01621459.2015.1054487. URL http://dx.doi.org/10.1080/01621459.2015.1054487
-
[42]
An annealed sequential M onte C arlo method for B ayesian phylogenetics
Wang, L., Wang, S., and Bouchard-C \^o t \'e , A. An annealed sequential M onte C arlo method for B ayesian phylogenetics. Systematic Biology, 69 0 (1): 0 155--183, 2020
work page 2020
-
[43]
Wang, S. and Wang, L. Particle gibbs sampling for bayesian phylogenetic inference. Bioinformatics, 37 0 (5): 0 642--649, 10 2020. ISSN 1367-4803. doi:10.1093/bioinformatics/btaa867. URL https://doi.org/10.1093/bioinformatics/btaa867
-
[44]
Xie, T. and Zhang, C. ART ree: A deep autoregressive model for phylogenetic inference. In Thirty-seventh Conference on Neural Information Processing Systems, 2023. URL https://openreview.net/forum?id=SoLebIqHgZ
work page 2023
-
[45]
Xie, T., Richman, H., Gao, J., Matsen Iv, F. A., and Zhang, C. Phylovae: Unsupervised learning of phylogenetic trees via variational autoencoders. 2025
work page 2025
-
[46]
Improved variational bayesian phylogenetic inference with normalizing flows
Zhang, C. Improved variational bayesian phylogenetic inference with normalizing flows. In Proceedings of the 34th International Conference on Neural Information Processing Systems, NIPS '20, Red Hook, NY, USA, 2020. Curran Associates Inc. ISBN 9781713829546
work page 2020
- [47]
- [48]
-
[49]
Zhou, Y., Johansen, A. M., and Aston, J. A. Toward automatic model comparison: an adaptive sequential M onte C arlo approach. Journal of Computational and Graphical Statistics, 25 0 (3): 0 701--726, 2016
work page 2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.