Recognition: 2 theorem links
· Lean TheoremScalable vertex guided filtrations identify structurally relevant genes in cancer networks
Pith reviewed 2026-05-13 00:56 UTC · model grok-4.3
The pith
Vertex function-based filtrations recover the same essential cancer genes as Vietoris-Rips while scaling to larger networks and revealing higher-order cavities.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
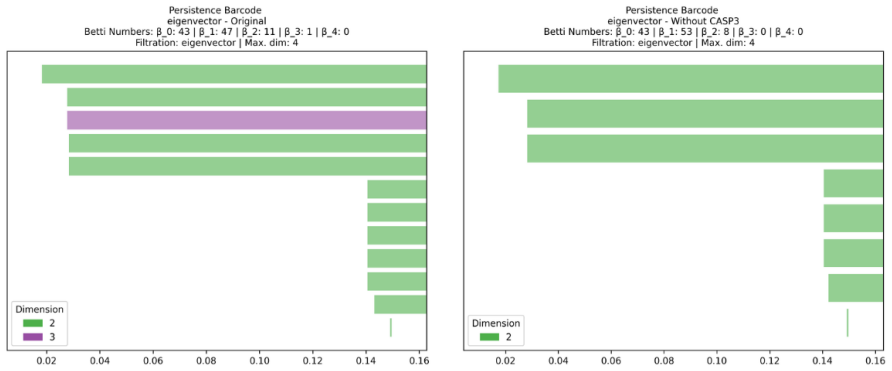
The authors establish that vertex function-based filtrations constructed from ordinary network measures produce persistent homology barcodes whose second Betti numbers match those of Vietoris-Rips filtrations on cancer-associated protein networks, thereby recovering known essential genes, nominating additional driver genes validated in IntOGen and NCG, and enabling the computation of third-order features that were previously inaccessible.
What carries the argument
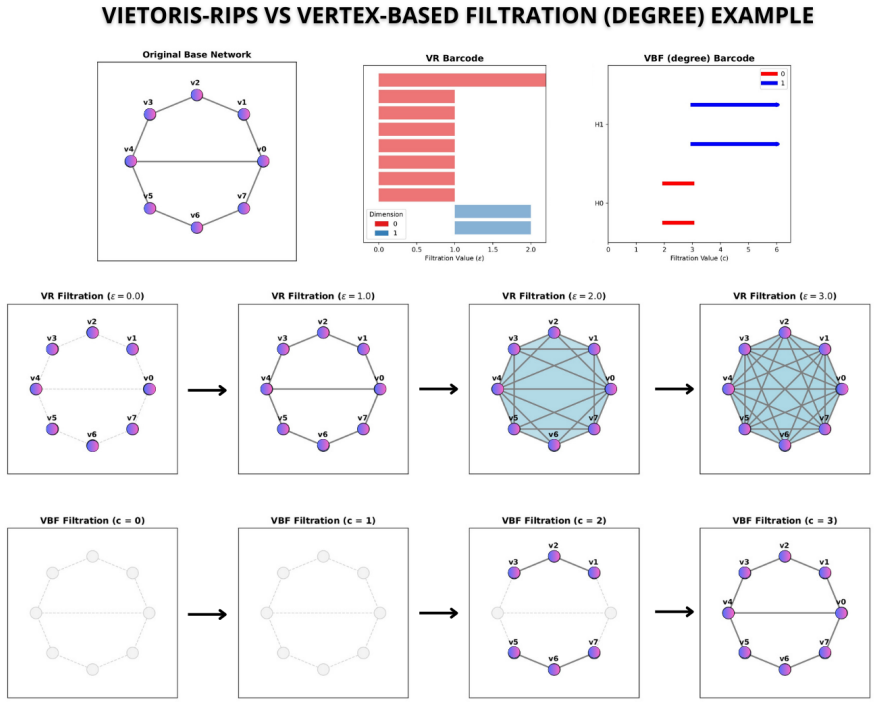
vertex function-based (VFB) filtration, which adds simplices in the order determined by a vertex function derived from network measures rather than from pairwise distances
If this is right
- Cancer networks can be screened for higher-dimensional holes without prohibitive computation.
- New candidate driver genes can be proposed on the basis of their contribution to persistent cycles or cavities.
- Classical network measures acquire a direct topological interpretation when used to guide filtrations.
- Topological gene prioritization becomes practical on networks too large for full Vietoris-Rips.
Where Pith is reading between the lines
- If the same vertex functions preserve topology across multiple cancer types, the method could serve as a standard preprocessing step for topological analysis of large interactomes.
- Genes highlighted by persistent higher-order features may mark points whose removal disrupts global connectivity in ways not captured by degree or betweenness alone.
- Experimental tests could check whether knocking out the newly identified genes alters measured Betti numbers in cellular models.
Load-bearing premise
That ordering vertices by common network measures produces the same persistent topological features as the distance-based Vietoris-Rips filtration.
What would settle it
A side-by-side computation on the same cancer network in which the birth and death times of Betti-2 features differ substantially between VFB and Vietoris-Rips, or in which the newly nominated genes show no enrichment for functional roles beyond random selection.
Figures

read the original abstract
Topological data analysis (TDA) has established itself as a useful tool for capturing multiscale structures in complex networks, such as connected components, cycles, and cavities. Although Vietoris-Rips (VR) filtering is widely used in network analysis, it tends to be computationally expensive, especially for large networks. This work explores vertex function-based (VFB) filtering based on network measures, applying persistent homology to identify relevant topological structures in cancer-associated protein networks, and compares its effectiveness with the VR approach. The results show that VFB reproduces the second-order structures (Betti-2) identified by VR, recovering previously reported essential genes. In addition, VFB detected new driver genes, confirmed in databases such as IntOGen and NCG, and allowed analysis of third-order structures (Betti-3) that was not feasible with VR. Thus, VFB represents a scalable alternative to VR, preserving biological interpretability and complementing classical network metrics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes vertex function-based (VFB) filtrations, derived from standard network centrality measures, as a computationally scalable alternative to Vietoris-Rips (VR) filtrations for computing persistent homology in large cancer-associated protein interaction networks. It claims that VFB reproduces the second Betti numbers and associated structures identified by VR, recovers known essential genes, identifies novel driver genes validated against IntOGen and NCG databases, and enables the computation of third-order topological features (Betti-3) that were infeasible with VR due to computational cost.
Significance. If the central claims hold, this work would offer a practical method for applying topological data analysis to large-scale biological networks, potentially uncovering multiscale structural features linked to cancer biology that complement traditional network metrics. The ability to access higher-order homology groups like Betti-3 could open new avenues for identifying complex topological motifs in disease networks. However, the significance is limited by the absence of rigorous quantitative comparisons between VFB and VR persistence diagrams and by the reliance on database overlap for validation, which may not isolate the contribution of the topological analysis.
major comments (4)
- Abstract and Results: The claim that VFB reproduces Betti-2 structures identified by VR lacks supporting quantitative evidence such as bottleneck distances, Wasserstein distances, or explicit cycle matching between the two persistence diagrams. Overlap in recovered gene lists alone does not establish that the filtrations capture equivalent multiscale topological features.
- Validation section: Confirmation of new driver genes in IntOGen and NCG databases shows cancer association but does not test whether the persistent homology signal provides information beyond the vertex function (e.g., degree or betweenness centrality) itself. A control analysis applying the same network measure without the filtration step is needed to establish added topological value.
- Methods: No details are given on the choice of specific vertex functions, sensitivity to their definitions, or how sublevel sets are constructed for the VFB. This omission makes it impossible to evaluate reproducibility or to determine whether the approach is parameter-free.
- Betti-3 results: While VFB permits computation of third-order features unavailable to VR, the manuscript provides no external biological validation, comparison to known cancer-related structures, or checks against filtration artifacts for these higher Betti numbers.
minor comments (2)
- The abstract would be strengthened by including at least one quantitative metric (e.g., overlap percentage or statistical test) for the Betti-2 reproduction and gene recovery claims.
- Figure legends and methods should explicitly state the network measures used as vertex functions and any preprocessing steps applied to the protein interaction networks.
Simulated Author's Rebuttal
We thank the referee for their insightful comments on our manuscript. We have carefully considered each point and made revisions to address the concerns, including adding quantitative comparisons and methodological clarifications. Our responses to the major comments are as follows.
read point-by-point responses
-
Referee: Abstract and Results: The claim that VFB reproduces Betti-2 structures identified by VR lacks supporting quantitative evidence such as bottleneck distances, Wasserstein distances, or explicit cycle matching between the two persistence diagrams. Overlap in recovered gene lists alone does not establish that the filtrations capture equivalent multiscale topological features.
Authors: We agree that quantitative metrics would strengthen the claim of reproduction. In the revised manuscript, we now include bottleneck and Wasserstein distances between the VFB and VR persistence diagrams for Betti-2, demonstrating their similarity. We also provide examples of matched cycles to show structural equivalence beyond gene list overlap. revision: yes
-
Referee: Validation section: Confirmation of new driver genes in IntOGen and NCG databases shows cancer association but does not test whether the persistent homology signal provides information beyond the vertex function (e.g., degree or betweenness centrality) itself. A control analysis applying the same network measure without the filtration step is needed to establish added topological value.
Authors: To address this, we have performed a control analysis in the revision where genes are ranked solely by the vertex function values without applying the filtration and persistent homology. We compare the overlap with known drivers and show that the topological features from VFB identify additional relevant genes not captured by the centrality measures alone, thus demonstrating the added value of the multiscale analysis. revision: yes
-
Referee: Methods: No details are given on the choice of specific vertex functions, sensitivity to their definitions, or how sublevel sets are constructed for the VFB. This omission makes it impossible to evaluate reproducibility or to determine whether the approach is parameter-free.
Authors: We have expanded the Methods section to include detailed descriptions of the vertex functions used (degree, betweenness, and eigenvector centrality), the construction of sublevel sets by sorting vertices according to the function values and building the filtration accordingly, and a sensitivity analysis showing robustness to small variations in function definitions. The method relies on standard network measures and is parameter-free in the sense that no additional thresholds are introduced beyond the choice of the measure. revision: yes
-
Referee: Betti-3 results: While VFB permits computation of third-order features unavailable to VR, the manuscript provides no external biological validation, comparison to known cancer-related structures, or checks against filtration artifacts for these higher Betti numbers.
Authors: We acknowledge the need for validation of Betti-3 features. In the revised version, we have added comparisons of the identified Betti-3 structures to known cancer-related pathways and modules from the literature, as well as robustness checks by perturbing the network slightly and re-computing to assess artifact sensitivity. However, comprehensive database validation for higher-order topological features is limited, and we discuss this as a direction for future research. revision: partial
Circularity Check
No significant circularity; empirical comparison and external validation are independent of inputs
full rationale
The paper applies vertex-function-based filtrations (using standard network measures) to cancer protein networks and compares the resulting persistent homology (Betti numbers) to Vietoris-Rips filtrations. It reports reproduction of Betti-2 structures, recovery of known essential genes, discovery of additional genes validated against IntOGen and NCG databases, and extension to Betti-3. No equations, parameter-fitting steps, self-definitional constructions, or load-bearing self-citations appear in the abstract or described content. The central claims rest on direct computational output and external database lookup rather than any reduction of results to the method's own definitions or fitted values. The derivation chain is therefore self-contained and non-circular.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Persistent homology applied to network filtrations captures biologically meaningful multiscale structures such as connected components, cycles, and cavities.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking unclearvertex-function-based (VFB) filtration is induced by a function g:V→R, extended to simplices by f(σ)=max g(v) (sublevel filtration)
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearVFB reproduces the second-order structures (Betti-2) identified by VR
Reference graph
Works this paper leans on
-
[1]
Gene regulatory networks analysis for the discovery of prognostic genes in gliomas
BARCELOS, Pedro Marçal et al. Gene regulatory networks analysis for the discovery of prognostic genes in gliomas. Scientific Reports, v. 15, n. 1, p. 14034, 2025
work page 2025
-
[2]
POP, Romana T. et al. Gene regulatory network integration with multi-omics data enhances survival predictions in cancer. Briefings in Bioinformatics, v. 26, n. 4, p. bbaf315, 2025
work page 2025
-
[3]
Wu, G., Feng, X., & Stein, L. (2010). A human functional protein interaction network and its application to cancer data analysis. Genome biology, 11(5), R53
work page 2010
-
[4]
C., Porras, P., Aranda, B., Hermjakob, H., & Orchard, S
Koh, G. C., Porras, P., Aranda, B., Hermjakob, H., & Orchard, S. E. (2012). Analyzing protein–protein interaction networks. Journal of proteome research, 11(4), 2014-2031
work page 2012
-
[5]
Ramos, R. H., Ferreira, C. D. O. L., & Simao, A. (2024). Human protein–protein interaction networks: A topological comparison review. Heliyon, 10(5)
work page 2024
-
[6]
NICOLAU, Monica; LEVINE, Arnold J.; CARLSSON, Gunnar. Topology based data anal- ysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proceedings of the National Academy of Sciences, v. 108, n. 17, p. 7265-7270, 2011
work page 2011
-
[7]
Applications of topological data analysis in oncology
BUKKURI, Anuraag; ANDOR, Noemi; DARCY, Isabel K. Applications of topological data analysis in oncology. Frontiers in artificial intelligence, v. 4, p. 659037, 2021
work page 2021
-
[8]
Identifying key genes in cancer networks using persistent homology
RAMOS, Rodrigo Henrique et al. Identifying key genes in cancer networks using persistent homology. Scientific Reports, v. 15, n. 1, p. 2751, 2025. 11
work page 2025
-
[9]
A roadmap for the computation of persistent homology
OTTER, Nina et al. A roadmap for the computation of persistent homology. EPJ Data Science, v. 6, n. 1, p. 17, 2017
work page 2017
-
[10]
Revisiting the use of graph centrality models in biolog- ical pathway analysis
NADERI YEGANEH, Pourya et al. Revisiting the use of graph centrality models in biolog- ical pathway analysis. BioData mining, v. 13, n. 1, p. 5, 2020
work page 2020
-
[11]
Topological data analysis for genomics and evolution: topology in biology
RABADÁN, Raúl; BLUMBERG, Andrew J. Topological data analysis for genomics and evolution: topology in biology. Cambridge University Press, 2019
work page 2019
-
[12]
LOUGHREY, Ciara F. et al. The topology of data: opportunities for cancer research. Bioin- formatics, v. 37, n. 19, p. 3091-3098, 2021
work page 2021
-
[13]
Computational topology: an introduction
EDELSBRUNNER, Herbert; HARER, John. Computational topology: an introduction. American Mathematical Soc., 2010
work page 2010
-
[14]
ZOMORODIAN,Afra.FastconstructionoftheVietoris-Ripscomplex.Computers&Graph- ics, v. 34, n. 3, p. 263-271, 2010
work page 2010
-
[15]
Persistence homology of net- works: methods and applications
AKTAS, Mehmet E.; AKBAS, Esra; FATMAOUI, Ahmed El. Persistence homology of net- works: methods and applications. Applied Network Science, v. 4, n. 1, p. 1-28, 2019
work page 2019
-
[16]
Project, T. G. GUDHIUser and Reference Manual(GUDHI Editorial Board, 2024), 3.10.1 edn
work page 2024
-
[17]
BARABÁSI, Albert-László. Network science. Philosophical Transactions of the Royal Soci- ety A: Mathematical, Physical and Engineering Sciences, v. 371, n. 1987, 2013
work page 1987
-
[18]
Major apoptotic mechanisms and genes involved in apoptosis
Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016 Jul;37(7):8471-86
work page 2016
-
[19]
GUTTÀ, Cristiano et al. Low expression of pro-apoptotic proteins Bax, Bak and Smac indicates prolonged progression-free survival in chemotherapy-treated metastatic melanoma. Cell death & disease, v. 11, n. 2, p. 124, 2020
work page 2020
-
[20]
MANN, Jasdeep et al. Non-canonical BAD activity regulates breast cancer cell and tumor growth via 14-3-3 binding and mitochondrial metabolism. Oncogene, v. 38, n. 18, p. 3325- 3339, 2019
work page 2019
-
[21]
BCL-2 and BCL-xL in Cancer: Regulation, Function, and Thera- peutic Targeting
SILVA, João PN et al. BCL-2 and BCL-xL in Cancer: Regulation, Function, and Thera- peutic Targeting. International Journal of Molecular Sciences, v. 27, n. 2, p. 1123, 2026
work page 2026
-
[22]
Current and Emerging Therapies for Targeting the ERK1/2 & PI3K Pathways in Cancer
ABIZADEH, Ethan et al. Current and Emerging Therapies for Targeting the ERK1/2 & PI3K Pathways in Cancer. International Journal of Molecular Sciences, v. 26, n. 17, p. 8696, 2025
work page 2025
-
[23]
Wu, Q., Wu, W., Fu, B., Shi, L., Wang, X., & Kuca, K. (2019). JNK signaling in cancer cell survival. Medicinal research reviews, 39(6), 2082-2104
work page 2019
-
[24]
SOENGAS, María S. et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature, v. 409, n. 6817, p. 207-211, 2001
work page 2001
-
[25]
FUTAMI, Kazunobu; FURUICHI, Yasuhiro. RECQL1 and WRN DNA repair helicases: potential therapeutic targets and proliferative markers against cancers. Frontiers in genetics, v. 5, p. 441, 2015
work page 2015
-
[26]
ZUO, Chunjian et al. Polymorphisms in ERCC4 and ERCC5 and risk of cancers: Systematic research synopsis, meta-analysis, and epidemiological evidence. Frontiers in oncology, v. 12, p. 951193, 2022. 12
work page 2022
-
[27]
CONCEICAO, Carlota JF et al. PARP1: A comprehensive review of its mechanisms, thera- peutic implications and emerging cancer treatments. Biochimica et Biophysica Acta (BBA)- Reviews on Cancer, v. 1880, n. 2, p. 189282, 2025
work page 2025
-
[28]
The functions of PCNA in tumor stemness and invasion
WANG, Yuan-Liang et al. The functions of PCNA in tumor stemness and invasion. Inter- national Journal of Molecular Sciences, v. 23, n. 10, p. 5679, 2022
work page 2022
-
[29]
JO, Sora et al. O-GlcNAcylation of DDB1 at Ser-764 by OGT promotes cancer cell stemness in colorectal cancer through increased polyubiquitination of p53. Scientific reports, 2025
work page 2025
-
[30]
Mutant p53 in cancer: from molecular mechanism to therapeutic modulation
CHEN, Xiaohua et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell death & disease, v. 13, n. 11, p. 974, 2022
work page 2022
-
[31]
WEBER-LASSALLE, Nana et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Research, v. 21, n. 1, p. 55, 2019. 13
work page 2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.