0
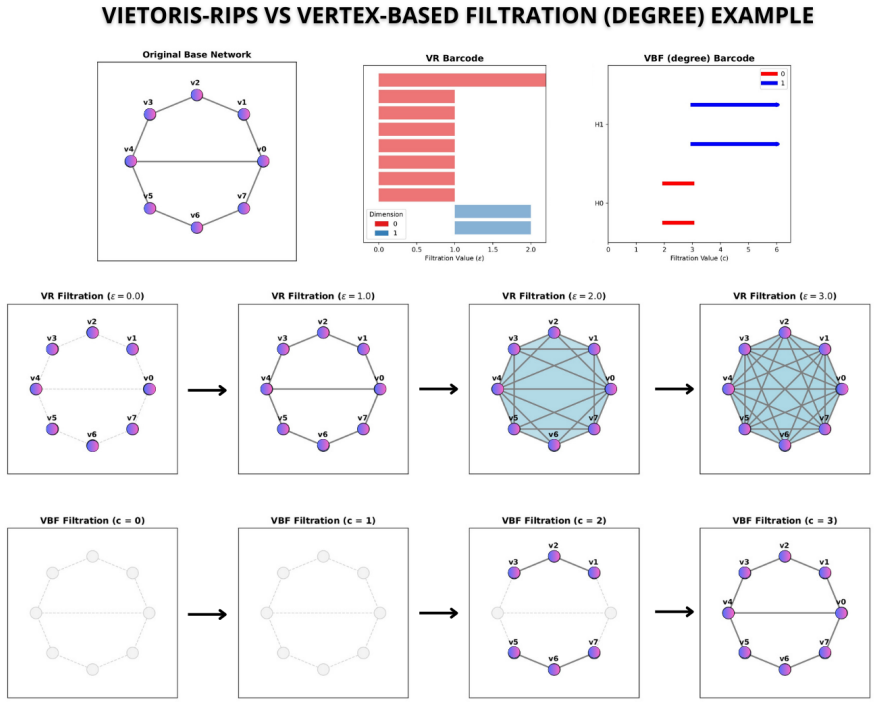
Vertex measures match VR in spotting cancer genes
Scalable vertex guided filtrations identify structurally relevant genes in cancer networks
They recover known essentials, nominate new drivers, and compute cavities where full VR fails.
 full image
full image
abstract click to expand
Topological data analysis (TDA) has established itself as a useful tool for capturing multiscale structures in complex networks, such as connected components, cycles, and cavities. Although Vietoris-Rips (VR) filtering is widely used in network analysis, it tends to be computationally expensive, especially for large networks. This work explores vertex function-based (VFB) filtering based on network measures, applying persistent homology to identify relevant topological structures in cancer-associated protein networks, and compares its effectiveness with the VR approach. The results show that VFB reproduces the second-order structures (Betti-2) identified by VR, recovering previously reported essential genes. In addition, VFB detected new driver genes, confirmed in databases such as IntOGen and NCG, and allowed analysis of third-order structures (Betti-3) that was not feasible with VR. Thus, VFB represents a scalable alternative to VR, preserving biological interpretability and complementing classical network metrics.