Deviance-style normalization for jointly overdispersed counts
Pith reviewed 2026-06-25 19:16 UTC · model grok-4.3
The pith
Dirichlet-multinomial deviance residuals normalize sparse jointly overdispersed counts while preserving exact zeros and recovering the multinomial limit.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the Dirichlet-multinomial deviance residualization preserves exact sparsity, evaluates in constant time per nonzero entry, agrees with multinomial residuals on singleton counts, shrinks repeated-count residuals according to the overdispersion the null tolerates, and recovers the multinomial residual as the concentration parameter tends to infinity; the same fixed-dispersion comparison principle extends to ordered and tree-structured features via the generalized DM and the Dirichlet-tree multinomial, yielding a single residual family that subsumes joint and feature-wise count nulls.
What carries the argument
The Dirichlet-multinomial deviance residual, obtained by comparing each observed count to its conditional expectation under a fixed-total, single-concentration null formed by conditioning independent negative binomials.
If this is right
- Sparse count pipelines can apply joint overdispersion correction without densifying the matrix or changing asymptotic cost.
- The residuals apply unchanged to data whose categories are ordered or arranged in a tree via the generalized and Dirichlet-tree models.
- Feature-wise negative-binomial models and ordinary multinomial models become limiting cases of one common residual construction.
- Normalization remains valid on exactly sparse data because no artificial nonzeros are introduced.
Where Pith is reading between the lines
- The construction supplies a uniform way to produce comparable residuals across experiments that differ only in their total sequencing depth.
- Downstream tasks such as clustering or differential testing could be rerun with these residuals to test whether joint overdispersion adjustment changes biological conclusions.
- Analogous conditioning arguments might yield deviance residuals for other families of count distributions that admit a fixed-total interpretation.
Load-bearing premise
Each sample's count vector behaves as independent negative-binomial draws conditioned on their observed total, with one shared scalar concentration governing all overdispersion.
What would settle it
Generate data from independent negative binomials conditioned on observed totals and check whether the proposed residuals leave singleton counts unchanged while shrinking the magnitude of residuals for counts greater than one exactly in line with the chosen concentration value.
Figures
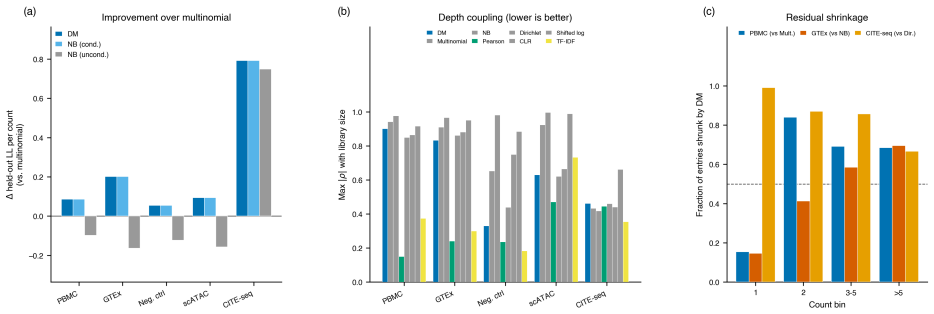
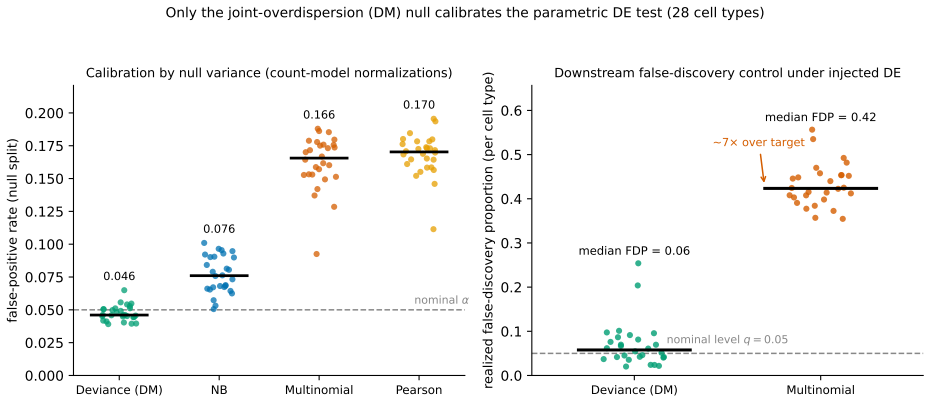




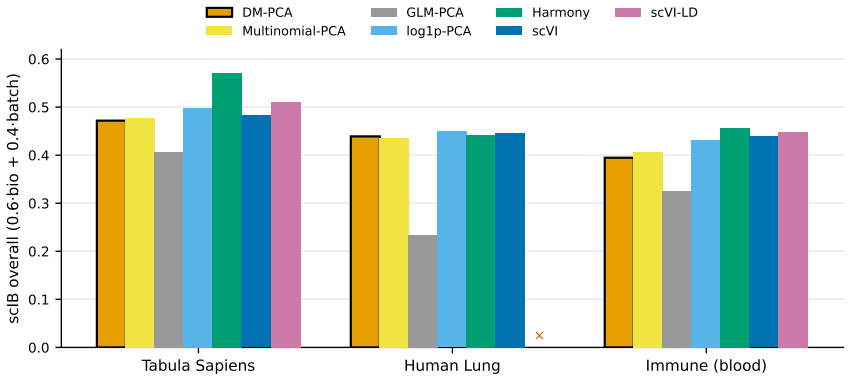










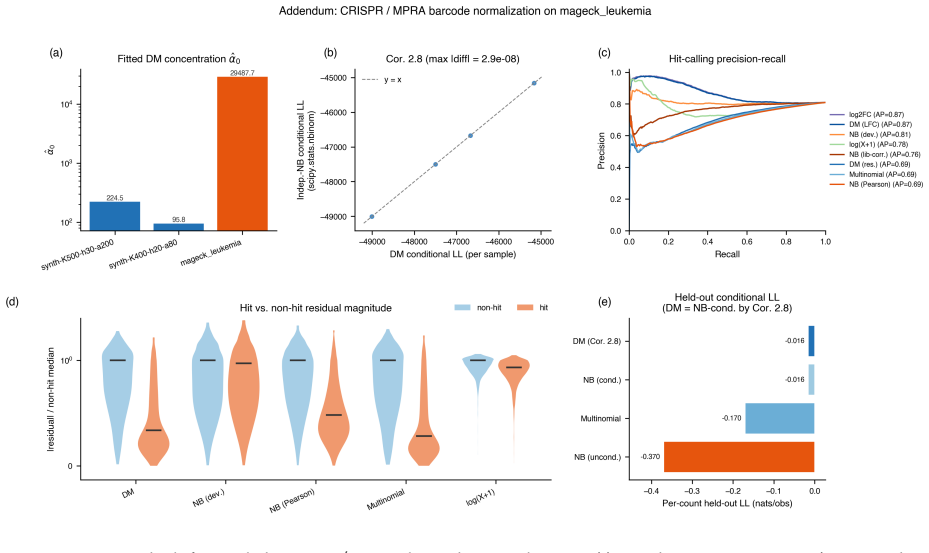

read the original abstract
We introduce a Dirichlet--multinomial (DM) deviance residualization for sparse, jointly overdispersed count matrices, the regime that dominates sequencing-based biochemical assays. The DM null treats each sample's count vector as a fixed-total composition with a single scalar concentration $\alpha_0$ governing overdispersion, and arises exactly by conditioning independent negative-binomial feature counts on the observed sample total -- making the DM the joint conditional analogue of standard feature-wise overdispersed count models. The resulting transform preserves exact sparsity, evaluates in constant time per nonzero entry, agrees with multinomial residuals on singleton counts, shrinks repeated-count residuals according to the overdispersion the null tolerates, and recovers the multinomial residual as $\alpha_0\to\infty$. The same fixed-dispersion comparison principle extends to ordered and tree-structured features via the generalized DM and the Dirichlet-tree multinomial, giving a single residual family that subsumes joint and feature-wise count nulls under a common compositional logic and is computationally lightweight enough to drop into existing sparse pipelines.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a Dirichlet-multinomial (DM) deviance residualization for sparse, jointly overdispersed count matrices common in sequencing assays. The DM null is defined as a fixed-total composition with scalar concentration α0, obtained exactly by conditioning independent negative-binomial feature counts on the observed sample total. The resulting residual is claimed to preserve exact sparsity, evaluate in O(1) time per nonzero entry, agree with multinomial residuals on singleton counts, shrink repeated-count residuals according to tolerated overdispersion, recover the multinomial residual as α0→∞, and extend via the same fixed-dispersion principle to generalized DM and Dirichlet-tree multinomial models for ordered or tree-structured features.
Significance. If the derivation and claimed properties hold, the method would supply a computationally lightweight, sparsity-preserving normalization that unifies joint compositional and feature-wise overdispersed count models under a common conditional logic, with direct applicability to high-throughput biochemical count data.
major comments (1)
- Abstract: The manuscript states conceptual properties and the derivation route (conditioning negative binomials on totals) but supplies no equations, explicit residual formula, proofs, simulations, or data examples; therefore the mathematical support for the central claims (sparsity preservation, O(1) evaluation, shrinkage behavior, and limit recovery) cannot be verified from the available information.
Simulated Author's Rebuttal
We thank the referee for the review and recommendation. We address the major comment below.
read point-by-point responses
-
Referee: [—] Abstract: The manuscript states conceptual properties and the derivation route (conditioning negative binomials on totals) but supplies no equations, explicit residual formula, proofs, simulations, or data examples; therefore the mathematical support for the central claims (sparsity preservation, O(1) evaluation, shrinkage behavior, and limit recovery) cannot be verified from the available information.
Authors: We agree that the abstract is intentionally high-level and narrative, without explicit equations or proofs, due to typical length constraints. The full manuscript supplies the explicit DM deviance residual formula (derived exactly by conditioning independent negative-binomial counts on the observed sample total), the proofs of sparsity preservation and O(1) evaluation per nonzero entry, the recovery of the multinomial residual as α₀→∞, the shrinkage behavior under finite α₀, and the supporting simulation studies and data examples. To address the concern about verifiability from the abstract, we will revise it to include a concise statement of the key residual formula. revision: yes
Circularity Check
No significant circularity identified
full rationale
The manuscript presents the DM null and its deviance residual as following directly from the standard statistical construction of the Dirichlet-multinomial distribution (conditioning independent negative-binomial counts on their sum). All listed properties—sparsity preservation, O(1) evaluation, agreement with multinomial residuals at count=1, shrinkage under overdispersion, and the α0→∞ limit—are algebraic consequences of that construction plus the definition of deviance residuals under a fixed-α0 null. No load-bearing step reduces to a fitted parameter renamed as a prediction, a self-citation chain, or an ansatz smuggled via prior work by the same authors. The extension to generalized DM and Dirichlet-tree multinomial applies the identical fixed-dispersion comparison principle to the corresponding joint distributions. The derivation is therefore self-contained against external benchmarks and receives the default non-circularity finding.
Axiom & Free-Parameter Ledger
free parameters (1)
- α0
axioms (1)
- domain assumption The DM distribution arises exactly by conditioning independent negative-binomial feature counts on the observed sample total.
Reference graph
Works this paper leans on
-
[1]
Comparison of transformations for single-cell RNA-seq data
Constantin Ahlmann-Eltze and Wolfgang Huber. Comparison of transformations for single-cell RNA-seq data. Na- ture Methods, 20(5):665–672, 2023
2023
-
[2]
Comparison of transformations for single-cell RNA-seq data
Constantin Ahlmann-Eltze and Wolfgang Huber. Comparison of transformations for single-cell RNA-seq data. Na- ture Methods, 20(5):665–672, 2023. Alias of AhlmannEltzeHuber2023
2023
-
[3]
The statistical analysis of compositional data
John Aitchison. The statistical analysis of compositional data. Journal of the Royal Statistical Society: Series B , 44(2): 139–160, 1982
1982
-
[4]
The Statistical Analysis of Compositional Data
John Aitchison. The Statistical Analysis of Compositional Data. Chapman & Hall, 1986
1986
-
[5]
John Aitchison and C. H. Ho. The multivariate Poisson-log normal distribution. Biometrika, 76(4):643–653, 1989
1989
-
[6]
John Aitchison and S. M. Shen. Logistic-normal distributions: Some properties and uses. Biometrika, 67(2):261–272, 1980
1980
-
[7]
Differential expression analysis for sequence count data
Simon Anders and Wolfgang Huber. Differential expression analysis for sequence count data. Genome Biology , 11(10):R106, 2010. doi: 10.1186/gb-2010-11-10-r106. url-verified 2026-05-26: https://link.springer.com/article/10.1186/gb-2010-11-10-r106
-
[8]
Latent Dirichlet allocation
David M Blei, Andrew Y Ng, and Michael I Jordan. Latent Dirichlet allocation. Journal of Machine Learning Research, 3:993–1022, 2003
2003
-
[9]
Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2
Evan Bolyen, Jai Ram Rideout, Matthew R Dillon, Nicholas A Bokulich, Christian C Abnet, Gabriel A Al-Ghalith, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37 (8):852–857, 2019
2019
-
[10]
Mueller, Fabian J
Maren Buettner, Johannes Ostner, Christian L. Mueller, Fabian J. Theis, and Benjamin Schubert. scCODA is a Bayesian model for compositional single-cell data analysis. Nature Communications, 12(1):6876, 2021
2021
-
[11]
Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample
J Gregory Caporaso, Christian L Lauber, William A Walters, Donna Berg-Lyons, Catherine A Lozupone, Peter J Turn- baugh, Noah Fierer, and Rob Knight. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences , 108(Supplement 1):4516–4522, 2011
2011
-
[12]
Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis
Jun Chen and Hongzhe Li. Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis. The Annals of Applied Statistics, 7(1):418–442, 2013
2013
-
[13]
Comparison and evaluation of statistical error models for scRNA-seq
Saket Choudhary and Rahul Satija. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biology, 23(1):27, 2022. 63
2022
-
[14]
Connor and James E
Robert J. Connor and James E. Mosimann. Concepts of independence for proportions with a generalization of the dirichlet distribution. Journal of the American Statistical Association , 64(325):194–206, 1969
1969
-
[15]
Deep neural networks for YouTube recommendations
Paul Covington, Jay Adams, and Emre Sargin. Deep neural networks for YouTube recommendations. In Proceedings of the 10th ACM Conference on Recommender Systems (RecSys) , pages 191–198, 2016
2016
-
[16]
Performance of recommender algorithms on top- n recom- mendation tasks
Paolo Cremonesi, Yehuda Koren, and Roberto Turrin. Performance of recommender algorithms on top- n recom- mendation tasks. In Proceedings of the 4th ACM Conference on Recommender Systems (RecSys) , pages 39–46, 2010
2010
-
[17]
Sur les lois de probabilité à estimation exhaustive
Georges Darmois. Sur les lois de probabilité à estimation exhaustive. C. R. Acad. Sci. Paris, 260:1265–1268, 1935
1935
-
[18]
Samuel Y. Dennis. On the hyper-dirichlet type 1 and hyper-liouville distributions. Communications in Statistics— Theory and Methods, 20(12):4069–4081, 1991
1991
-
[19]
Exponential Families in Theory and Practice
Bradley Efron. Exponential Families in Theory and Practice. Institute of Mathematical Statistics T extbooks. Cambridge University Press, 2022. doi: 10.1017/9781108773157
-
[20]
Clustering documents with an exponential-family approximation of the dirichlet compound multi- nomial distribution
Charles Elkan. Clustering documents with an exponential-family approximation of the dirichlet compound multi- nomial distribution. In Proceedings of the 23rd International Conference on Machine Learning , pages 289–296, 2006
2006
-
[21]
Rouhana, Julia Waldman, et al
Gökcen Eraslan, Eugene Drokhlyansky, Shankara Anand, Evgenij Fiskin, Ayshwarya Subramanian, Michal Slyper, Jiali Wang, Nicholas Van Wittenberghe, John M. Rouhana, Julia Waldman, et al. Single-nucleus cross-tissue molec- ular reference maps toward understanding disease gene function. Science, 376(6594):eabl4290, 2022. Alias of EraslanDrokhlyanskyAnandEtAl2022
2022
-
[22]
Symmetric Multivariate and Related Distributions
Kai-Tai Fang. Symmetric Multivariate and Related Distributions. Chapman and Hall/CRC, 2018
2018
-
[23]
Farewell and Vernon T
Daniel M. Farewell and Vernon T. Farewell. Dirichlet negative multinomial regression for overdispersed correlated count data. Biostatistics, 14(2):395–404, 2013
2013
-
[24]
Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene se- quencing and selective growth experiments by compositional data analysis
Andrew D Fernandes, Jennifer NS Reid, Jean M Macklaim, Thomas A McMurrough, David R Edgell, and Gregory B Gloor. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene se- quencing and selective growth experiments by compositional data analysis. Microbiome, 2:15, 2014
2014
-
[25]
On the mathematical foundations of theoretical statistics
Ronald A Fisher. On the mathematical foundations of theoretical statistics. Philosophical transactions of the Royal Society of London. Series A, containing papers of a mathematical or physical character , 222(594-604):309–368, 1922
1922
-
[26]
Stochastic Finance: An Introduction in Discrete Time
Hans Föllmer and Alexander Schied. Stochastic Finance: An Introduction in Discrete Time . Walter de Gruyter, 3rd edition, 2011
2011
-
[27]
Gloor, Jean M
Gregory B. Gloor, Jean M. Macklaim, Vera Pawlowsky-Glahn, and Juan J. Egozcue. Microbiome datasets are com- positional: and this is not optional. Frontiers in Microbiology, 8:2224, 2017
2017
-
[28]
Lindrooth
Paulo Guimaraes and Richard C. Lindrooth. Controlling for overdispersion in grouped conditional logit models: a computationally simple application of dirichlet-multinomial regression. The Econometrics Journal, 10(2):439–452, 2007
2007
-
[29]
Gupta and Donald St P
Rameshwar D. Gupta and Donald St P. Richards. Multivariate liouville distributions. Journal of Multivariate Analysis, 23(2):233–256, 1987
1987
-
[30]
Normalization and variance stabilization of single-cell rna-seq data using regularized negative binomial regression
Christoph Hafemeister and Rahul Satija. Normalization and variance stabilization of single-cell rna-seq data using regularized negative binomial regression. Genome Biology, 20(1):296, 2019
2019
-
[31]
The MovieLens datasets: history and context
F Maxwell Harper and Joseph A Konstan. The MovieLens datasets: history and context. ACM Transactions on Inter- active Intelligent Systems, 5(4):1–19, 2015
2015
-
[32]
Harrison, W
Joshua G. Harrison, W. John Calder, Vivaswat Shastry, and C. Alex Buerkle. Dirichlet-multinomial modelling out- performs alternatives for analysis of microbiome and other ecological count data. Molecular Ecology Resources, 20(2): 481–497, 2020. 64
2020
-
[33]
BAGEL: a computational framework for identifying essential genes from pooled library screens
Traver Hart and Jason Moffat. BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics, 17:164, 2016
2016
-
[34]
Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens
Traver Hart, Amy Hin Yan T ong, Katie Chan, Jolanda Van Leeuwen, Ashwin Seetharaman, Michael Aregger, Megha Chandrashekhar, Nicole Hustedt, Sahil Seth, Avery Noonan, et al. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3: Genes, Genomes, Genetics , 7(8):2719–2727, 2017
2017
-
[35]
A closer look at the deviance
Trevor Hastie. A closer look at the deviance. The American Statistician, 41(1):16–20, 1987
1987
-
[36]
Hall, and Zvi Griliches
Jerry Hausman, Bronwyn H. Hall, and Zvi Griliches. Econometric models for count data with an application to the patents-r&d relationship. Econometrica, 52(4):909–938, 1984
1984
-
[37]
Howard, Aaditya Ramdas, Jon McAuliffe, and Jasjeet Sekhon
Steven R. Howard, Aaditya Ramdas, Jon McAuliffe, and Jasjeet Sekhon. Time-uniform, nonparametric, nonasymp- totic confidence sequences. The Annals of Statistics, 49(2):1055–1080, 2021. doi: 10.1214/20-AOS2002
-
[38]
Collaborative filtering for implicit feedback datasets
Yifan Hu, Yehuda Koren, and Chris Volinsky. Collaborative filtering for implicit feedback datasets. In Proceedings of the 8th IEEE International Conference on Data Mining (ICDM) , pages 263–272, 2008
2008
-
[39]
On distributions admitting a sufficient statistic
Bernard Osgood Koopman. On distributions admitting a sufficient statistic. Transactions of the American Mathemat- ical society, 39(3):399–409, 1936
1936
-
[40]
Koslovsky
Matthew D. Koslovsky. A bayesian zero-inflated dirichlet-multinomial regression model for multivariate composi- tional count data. Biometrics, 79(4):3239–3251, 2023
2023
-
[41]
Analytic Pearson residuals for normalization of single-cell RNA- seq UMI data
Jan Lause, Philipp Berens, and Dmitry Kobak. Analytic Pearson residuals for normalization of single-cell RNA- seq UMI data. Genome Biology , 22:258, 2021. doi: 10.1186/s13059-021-02451-7. url-verified 2026-05-26: https://link.springer.com/article/10.1186/s13059-021-02451-7
-
[42]
MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens
Wei Li, Han Xu, T engfei Xiao, Le Cong, Michael I Love, Feng Zhang, Rafael A Irizarry, Jun S Liu, Myles Brown, and X Shirley Liu. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biology, 15:554, 2014
2014
-
[43]
Love, Wolfgang Huber, and Simon Anders
Michael I. Love, Wolfgang Huber, and Simon Anders. Moderated estimation of fold change and dispersion for rna- seq data with deseq2. Genome Biology, 15(12):550, 2014
2014
-
[44]
Macosko, Anindita Basu, Rahul Satija, James Nemesh, Karthik Shekhar, Michael Goldman, Itay Tirosh, Al- lison R
Evan Z. Macosko, Anindita Basu, Rahul Satija, James Nemesh, Karthik Shekhar, Michael Goldman, Itay Tirosh, Al- lison R. Bialas, Nolan Kamitaki, Emily M. Martersteck, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5):1202–1214, 2015
2015
-
[45]
Marshall, Ingram Olkin, and Barry C
Albert W. Marshall, Ingram Olkin, and Barry C. Arnold. Inequalities: Theory of Majorization and Its Applications . Academic Press, 1979
1979
-
[46]
McCullagh and J
P. McCullagh and J. A. Nelder. Generalized Linear Models. Chapman and Hall, 2nd edition, 1989
1989
-
[47]
Thomas P. Minka. Estimating a dirichlet distribution. T echnical report, MIT, 2000
2000
-
[48]
Thomas P. Minka. The dirichlet-tree distribution. T echnical report, 2004. T echnical note, 1999; revised 2004
2004
-
[49]
Carl N. Morris. Natural exponential families with quadratic variance functions. The Annals of Statistics, 10(1):65–80, 1982
1982
-
[50]
Mosimann
James E. Mosimann. On the compound multinomial distribution, the multivariate β-distribution, and correlations among proportions. Biometrika, 49(1/2):65–82, 1962
1962
-
[51]
Mosimann
James E. Mosimann. On the compound negative multinomial distribution and correlations among inversely sampled pollen counts. Biometrika, 50(1/2):47–54, 1963
1963
-
[52]
Dirichlet and Related Distributions: Theory, Methods and Applica- tions
Kai-Wang Ng, Guo-Liang Tian, and Man-Lai Tang. Dirichlet and Related Distributions: Theory, Methods and Applica- tions. Wiley, 2011
2011
-
[53]
Sufficient statistics and intrinsic accuracy
Edwin James George Pitman. Sufficient statistics and intrinsic accuracy. InMathematical Proceedings of the cambridge Philosophical society, volume 32, pages 567–579. Cambridge University Press, 1936. 65
1936
-
[54]
Game-theoretic statistics and safe anytime-valid inference
Aaditya Ramdas, Peter Grünwald, Vladimir Vovk, and Glenn Shafer. Game-theoretic statistics and safe anytime-valid inference. Statistical Science, 38(4):576–601, 2023. doi: 10.1214/23-STS894
-
[55]
Using tf-idf to determine word relevance in document queries
Juan Ramos et al. Using tf-idf to determine word relevance in document queries. In Proceedings of the First Instruc- tional Conference on Machine Learning, volume 242, pages 29–48, 2003
2003
-
[56]
Robinson, Davis J
Mark D. Robinson, Davis J. McCarthy, and Gordon K. Smyth. edger: a bioconductor package for differential expres- sion analysis of digital gene expression data. Bioinformatics, 26(1):139–140, 2010
2010
-
[57]
T erm-weighting approaches in automatic text retrieval
Gerard Salton and Christopher Buckley. T erm-weighting approaches in automatic text retrieval. Information Pro- cessing & Management, 24(5):513–523, 1988
1988
-
[58]
Farrell, David Gennert, Alexander F
Rahul Satija, Jeffrey A. Farrell, David Gennert, Alexander F. Schier, and Aviv Regev. Spatial reconstruction of single- cell gene expression data. Nature Biotechnology, 33(5):495–502, 2015
2015
-
[59]
Fast mle computation for the dirichlet multinomial
Max Sklar. Fast mle computation for the dirichlet multinomial. arXiv preprint arXiv:1405.0099, 2014
arXiv 2014
-
[60]
Mauck III, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. Mauck III, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. Comprehensive integration of single- cell data. Cell, 177(7):1888–1902.e21, 2019. doi: 10.1016/j.cell.2019.05.031. url-verified 2026-05-26: https://www.cell.com/cell/fulltext/S0092-8674(19)30559-8
-
[61]
Droplet scrna-seq is not zero-inflated
Valentine Svensson. Droplet scrna-seq is not zero-inflated. Nature Biotechnology, 38(2):147–150, 2020
2020
-
[62]
Thorson, Kelli F
James T. Thorson, Kelli F. Johnson, Richard D. Methot, and Ian G. Taylor. Model-based estimates of effective sample size in stock assessment models using the dirichlet-multinomial distribution. Fisheries Research, 192:84–93, 2017
2017
-
[63]
Tiao and Irwin Cuttman
George C. Tiao and Irwin Cuttman. The inverted dirichlet distribution with applications. Journal of the American Statistical Association, 60(311):793–805, 1965
1965
-
[64]
F. William T ownes, Stephanie C. Hicks, Martin J. Aryee, and Rafael A. Irizarry. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biology , 20:295, 2019. doi: 10.1186/ s13059-019-1861-6. url-verified 2026-05-26: https://link.springer.com/article/10.1186/s13059-019-1861-6
-
[65]
Williams, Geo Pertea, Ali Mortazavi, Grace Kwan, Marijke J
Cole Trapnell, Brian A. Williams, Geo Pertea, Ali Mortazavi, Grace Kwan, Marijke J. van Baren, Steven L. Salzberg, Barbara J. Wold, and Lior Pachter. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28(5):511–515, 2010
2010
-
[66]
Duncan Wadsworth, Raffaele Argiento, Michele Guindani, Jessica Galloway-Peña, Samuel A
W. Duncan Wadsworth, Raffaele Argiento, Michele Guindani, Jessica Galloway-Peña, Samuel A. Shelburne, and Ma- rina Vannucci. An integrative bayesian dirichlet-multinomial regression model for the analysis of taxonomic abun- dances in microbiome data. BMC Bioinformatics, 18:94, 2017
2017
-
[67]
A dirichlet-tree multinomial regression model for associating dietary nutrients with gut microorganisms
Tao Wang and Hongyu Zhao. A dirichlet-tree multinomial regression model for associating dietary nutrients with gut microorganisms. Biometrics, 73(3):792–801, 2017
2017
-
[68]
Universal inference
Larry Wasserman, Aaditya Ramdas, and Sivaraman Balakrishnan. Universal inference. Proceedings of the National Academy of Sciences, 117(29):16880–16890, 2020
2020
-
[69]
A logistic normal multinomial regression model for microbiome compositional data analysis
Fengzhu Xia, Jun Chen, Wing Kam Fung, and Hongzhe Li. A logistic normal multinomial regression model for microbiome compositional data analysis. Biometrics, 69(4):1053–1063, 2013
2013
-
[70]
Mm algorithms for some discrete multivariate distributions.Journal of Computational and Graphical Statistics, 19(3):645–665, 2010
Hua Zhou and Kenneth Lange. Mm algorithms for some discrete multivariate distributions.Journal of Computational and Graphical Statistics, 19(3):645–665, 2010
2010
-
[71]
Nonparametric bayesian negative binomial factor analysis
Mingyuan Zhou. Nonparametric bayesian negative binomial factor analysis. Bayesian Analysis, 13(4):1065–1093, 2018. 66 Figure 4: Downstream analyses on GTEx. A deep generative baseline, scVI, is included wherever the comparison is commensurable. It attains the strongest embeddings here, but at far higher training cost and at the cost of differential- expre...
2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.