Smoothly Time-Varying Continuous Time Markov Chains in Phylogenetics
Pith reviewed 2026-06-26 00:29 UTC · model grok-4.3
The pith
A spline clock model represents log evolutionary rates as smooth cubic B-spline functions of time inside inhomogeneous CTMCs on phylogenies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The spline clock model parameterizes the log-transformed evolutionary rate as a smooth function of time using a cubic B-spline basis expansion in inhomogeneous continuous-time Markov chains acting along the branches of the phylogeny; integrals of the rate function over branches are approximated by Gauss-Legendre quadrature and smoothness is enforced by a Gaussian Markov random field prior on the spline coefficients.
What carries the argument
The spline clock model, which expands the log rate in cubic B-splines within an ICTMC and uses a GMRF prior plus quadrature for likelihoods.
If this is right
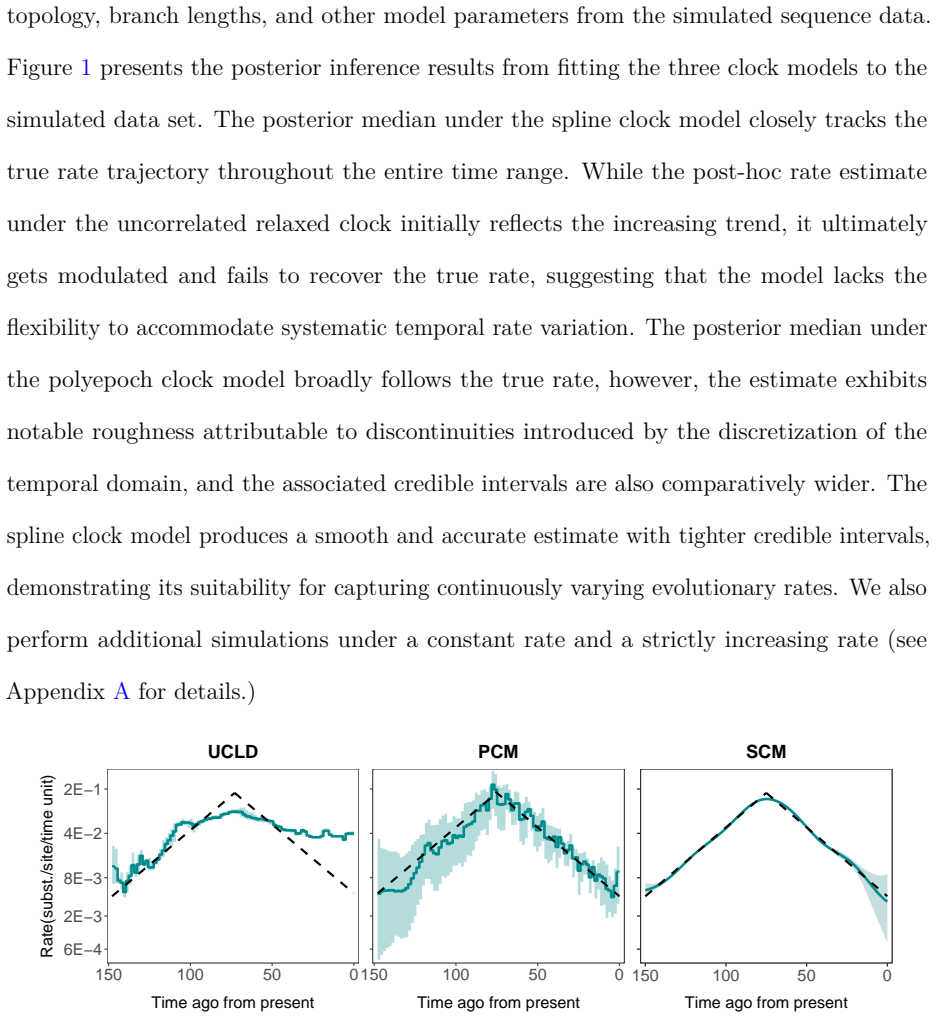
- Recovers true time-varying rates more accurately than competing clock models in simulations.
- Produces tighter credible intervals around the estimated rate trajectory.
- Detects strong time-varying signals in foamy virus substitution rates.
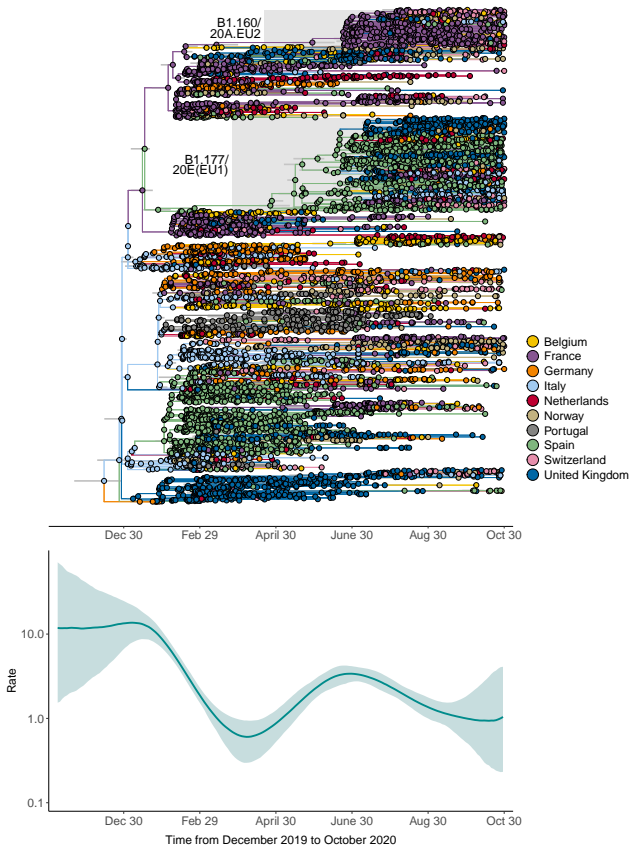
- Detects strong time-varying signals in SARS-CoV-2 spatial diffusion rates across Europe.
Where Pith is reading between the lines
- The same spline parameterization could be dropped into other CTMC-based models outside phylogenetics, such as continuous-time epidemic models.
- If smoothness is a reasonable default for many biological rate processes, the approach may reduce systematic bias in divergence-time estimates that assume constant rates.
- Datasets with documented sudden rate shifts (e.g., after host jumps or treatment changes) offer a direct test of when the smoothness prior becomes harmful.
Load-bearing premise
The log-transformed evolutionary rate can be adequately represented as a smooth function of time via cubic B-spline basis expansion.
What would settle it
A simulation study in which the true rate trajectory contains abrupt jumps or is otherwise non-smooth, after which the spline model shows larger error and wider intervals than a piecewise-constant or relaxed-clock alternative.
Figures
read the original abstract
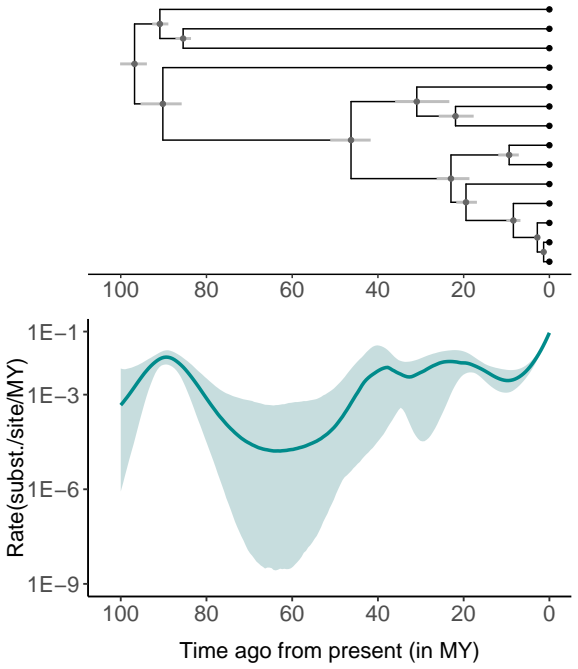
The dependence of evolutionary rate estimates on the timeframe of sampling poses a fundamental challenge for reconstructing evolutionary histories from molecular sequence data, which is central to evolutionary biology and infectious disease research. We present a novel and flexible approach to accommodate time-varying evolutionary rates by modeling the sequence substitution process using inhomogeneous continuous-time Markov chains (ICTMCs) acting along the branches of the phylogeny, and parameterizing the log transformed rate as a smooth function of time using a cubic B-spline basis expansion. Following the parlance of phylogenetics that refers to rates of molecular substitutions as molecular clocks, we call this a spline clock model. Integrals of the rate function over all branches, required for likelihood evaluation, are approximated efficiently using Gauss-Legendre quadrature, and smoothness is enforced by assigning a Gaussian Markov random field prior to the spline coefficients. Through a simulation study, we demonstrate that the spline clock model recovers the true time-varying rates more accurately and with tighter credible intervals than competing clock models. We apply the spline clock model to examine the evolutionary rate of foamy virus and the rate of spatial diffusion of SARS-CoV-2 across Europe, recovering strong time-varying signal in both settings.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the spline clock model, which parameterizes the log evolutionary rate as a smooth function of time via cubic B-spline basis expansion within an inhomogeneous continuous-time Markov chain (ICTMC) along phylogenetic branches. Smoothness is enforced via a Gaussian Markov random field (GMRF) prior on the spline coefficients, and branch integrals are approximated using Gauss-Legendre quadrature. The central claim is that a simulation study demonstrates more accurate recovery of true time-varying rates with tighter credible intervals than competing clock models; the model is then applied to foamy virus and SARS-CoV-2 data.
Significance. If the simulation results are robust, the approach supplies a flexible, internally consistent parameterization for time-varying rates that directly addresses a known challenge in molecular clock analyses. The modeling choices (B-splines on log-rate, GMRF prior, quadrature) are standard and do not introduce unsecured assumptions that would invalidate the likelihood or the recovery claim.
major comments (1)
- [Simulation study] The simulation study (referenced in the abstract and presumably detailed in the results section): the manuscript does not report the simulation design details required to evaluate the central claim, including how the true time-varying rates were generated, the specific competing clock models, number of replicates, data exclusion rules, or quantitative metrics for accuracy and interval width. This information is load-bearing for assessing whether the reported superior recovery holds.
minor comments (2)
- [Abstract] The abstract would benefit from a brief quantitative summary of the simulation metrics (e.g., error reduction or interval width ratios) to allow readers to gauge the strength of the central claim without reading the full results.
- [Methods] Clarify the exact number of quadrature points used in the Gauss-Legendre approximation and any sensitivity checks performed, even if the method is standard.
Simulated Author's Rebuttal
We thank the referee for their constructive review and recommendation of minor revision. We address the single major comment below regarding the simulation study.
read point-by-point responses
-
Referee: [Simulation study] The simulation study (referenced in the abstract and presumably detailed in the results section): the manuscript does not report the simulation design details required to evaluate the central claim, including how the true time-varying rates were generated, the specific competing clock models, number of replicates, data exclusion rules, or quantitative metrics for accuracy and interval width. This information is load-bearing for assessing whether the reported superior recovery holds.
Authors: We agree that the simulation study section requires additional detail to support the central claim. In the revised manuscript we will expand the Methods and Results sections to explicitly report: the procedure used to generate the true time-varying rates (including the specific functions or spline coefficients employed), the complete list of competing clock models against which performance was compared, the number of simulation replicates, any data exclusion or filtering rules applied, and the quantitative metrics used to assess accuracy and credible interval width (e.g., mean squared error, coverage, and average interval length). These additions will make the reported superior recovery fully evaluable. revision: yes
Circularity Check
No significant circularity
full rationale
The paper introduces a spline clock model parameterizing log-rate via cubic B-splines with GMRF prior and Gauss-Legendre quadrature for branch integrals in an inhomogeneous CTMC. The central claim is empirical recovery performance in a simulation study, which compares the fitted model against alternatives on held-out simulated data generated under known time-varying rates. This does not reduce to a self-definitional equivalence, fitted-input prediction, or self-citation chain; the simulation recovery metric is independent of the model's internal parameterization and prior choices. No load-bearing step equates a claimed result to its inputs by construction.
Axiom & Free-Parameter Ledger
free parameters (2)
- B-spline coefficients
- Basis expansion hyperparameters
axioms (1)
- domain assumption Sequence substitution along phylogeny branches can be modeled as an inhomogeneous continuous-time Markov chain with time-dependent rate.
Reference graph
Works this paper leans on
-
[1]
& Katzourakis, A
Aiewsakun, P. & Katzourakis, A. (2015), ‘Time dependency of foamy virus evolutionary rate estimates’,BMC Evolutionary Biology15,
2015
-
[2]
& Katzourakis, A
21 Aiewsakun, P. & Katzourakis, A. (2016), ‘Time-dependent rate phenomenon in viruses’, Journal of Virology90(16), 7184–7195. Aris-Brosou, S. & Yang, Z. (2003), ‘Bayesian models of episodic evolution support a late Precambrian explosive diversification of the Metazoa’,Molecular Biology and Evolution 20(12), 1947–1954. Baele, G., Carvalho, L. M., Brusselma...
2016
-
[3]
Bininda-Emonds, O. R. P., Cardillo, M., Jones, K. E., MacPhee, R. D. E., Beck, R. M. D., Grenyer, R., Price, S. A., Vos, R. A., Gittleman, J. L. & Purvis, A. (2007), ‘The delayed rise of present-day mammals’,Nature446(7135), 507–512. Brezger, A. & Lang, S. (2006), ‘Generalized structured additive regression based on Bayesian P-splines’,Computational Stati...
-
[4]
Duchêne, S., Holmes, E. C. & Ho, S. Y. (2014), ‘Analyses of evolutionary dynamics in viruses are hindered by a time-dependent bias in rate estimates’,Proceedings of the Royal Society B: Biological Sciences281(1786), 20140732. Dudas, G., Carvalho, L. M., Bedford, T., Tatem, A. J., Baele, G., Faria, N. R., Park, D. J., 23 Ladner, J. T., Arias, A., Asogun, D...
2014
-
[5]
J., Tristem, M., Gilbert, M
Katzourakis, A., Gifford, R. J., Tristem, M., Gilbert, M. T. P. & Pybus, O. G. (2009), ‘Macroevolution of complex retroviruses’,Science325(5947), 1512–1512. Kishino, H., Thorne, J. L. & Bruno, W. J. (2001), ‘Performance of a divergence time estimation method under a probabilistic model of rate evolution’,Molecular Biology and Evolution18(3), 352–361. 25 L...
2009
-
[6]
& Trivedi, K
Rindos, A., Woolet, S., Viniotis, I. & Trivedi, K. (1995), ‘Exact methods for the transient analysis for non-homogeneous continuous-time Markov chains’,Numerical Solutions of Markov Chains(NSMC)pp. 121–134. Rue, H. & Held, L. (2005),Gaussian Markov Random Fields: Theory and Applications, 1st edn, Chapman and Hall/CRC. Rue, H., Martino, S. & Chopin, N. (20...
1995
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.