Modeling Protein Evolution with Generative Models: from Extant Sequence Data to Evolutionary Dynamics
Pith reviewed 2026-06-30 01:48 UTC · model grok-4.3
The pith
Generative models trained on protein sequence families define probabilistic landscapes that can be coupled to population-genetic dynamics to simulate evolutionary trajectories.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
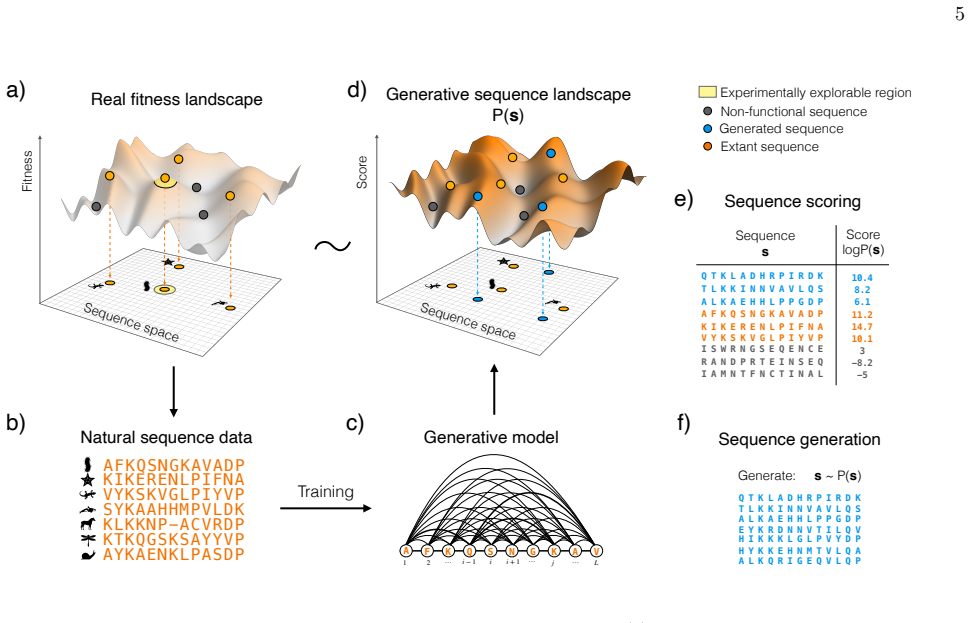
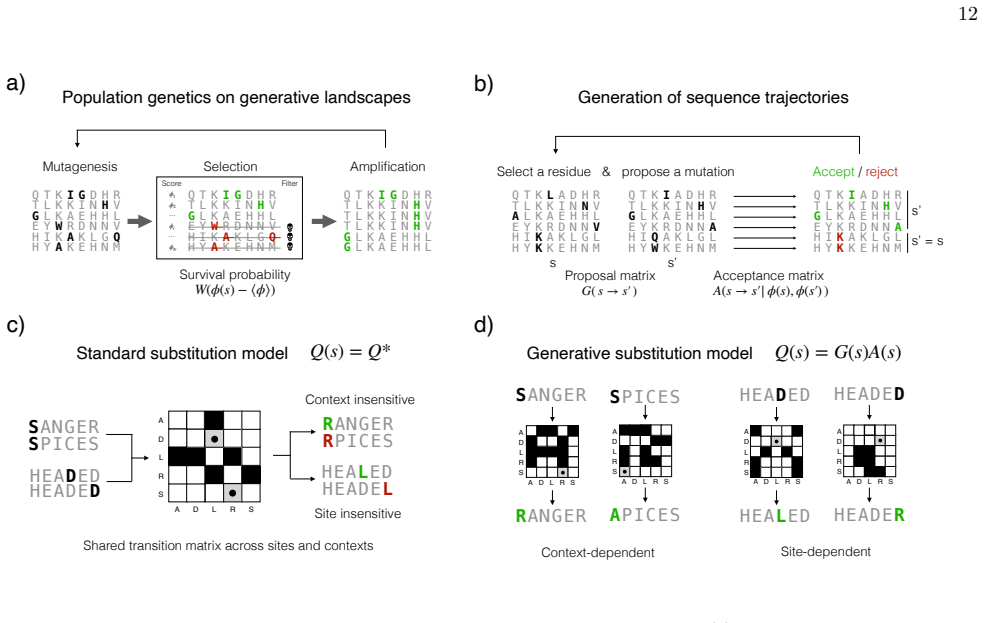
Generative sequence landscapes inferred from homologous families can be coupled to population-genetic or substitution-model dynamics to simulate protein evolution across experimental and phylogenetic timescales, with Direct Coupling Analysis serving as a validated, interpretable instance of the approach.
What carries the argument
Direct Coupling Analysis, which infers an energy function from multiple-sequence alignments to assign probabilities to entire sequences and thereby defines the landscape used for dynamics.
If this is right
- Simulations of viral evolution under changing selective pressures become feasible from sequence data alone.
- Laboratory drift experiments can be modeled in silico to predict reachable sequence variants.
- Historical contingency and entrenchment effects can be quantified by replaying evolution from different ancestral states.
- Epistatic contributions to substitution rates can be tracked over extended timescales.
- Long-term exploration of viable sequence space can be performed without enumerating all possible mutants.
Where Pith is reading between the lines
- The same coupling could be tested against codon-level mutation spectra to check whether the inferred landscapes remain predictive when mutation biases are included explicitly.
- Integration with structural data might reveal whether the landscapes implicitly encode three-dimensional constraints that are not captured by sequence statistics alone.
- If calibration between model scores and actual fitness improves, the framework could supply priors for forecasting which mutations are likely to fix in clinical or agricultural settings.
- Extension to include indels would require new generative architectures but could address a major gap in current sequence-space models.
Load-bearing premise
Landscapes fitted to present-day sequences are assumed to encode the functional constraints that governed past evolution and will govern future evolution without major distortion from sampling or model misspecification.
What would settle it
A direct comparison in which trajectories simulated from the coupled model systematically fail to reproduce the substitution patterns or fitness changes observed in a controlled laboratory evolution experiment or a well-resolved phylogenetic clade for the same protein.
Figures

read the original abstract
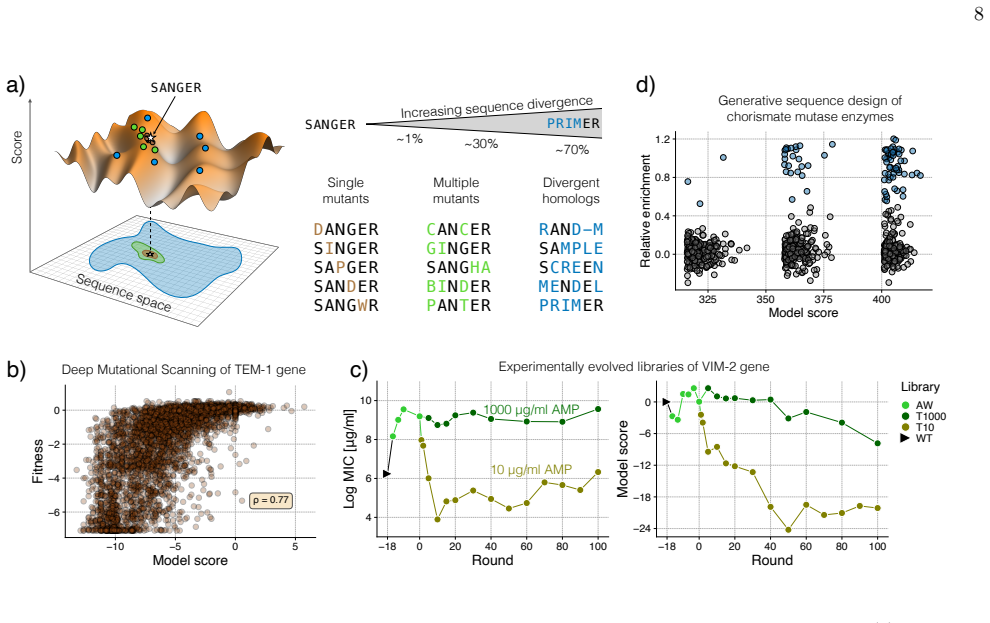
Protein sequences carry a record of evolutionary history shaped by mutation, selection, drift, and epistasis. Recent generative models trained on homologous sequence families offer a new way to read this record: they define probabilistic landscapes that score sequences, generate viable variants, and capture constraints that are difficult to measure experimentally. In this review, we discuss how such landscapes can be used not only for protein design or mutation-effect prediction, but also for modeling evolutionary dynamics. We focus particularly on Direct Coupling Analysis as an interpretable and experimentally validated framework, while placing it in the broader context of generative sequence modeling. We first describe how generative sequence landscapes are inferred and assessed, then review how they can be coupled to population-genetic or substitution-model dynamics to simulate protein evolution across experimental and phylogenetic timescales. Applications include viral evolution, laboratory drift experiments, historical contingency, entrenchment, epistatic drift over time, and long-term sequence-space exploration. We conclude by discussing open challenges, including score-fitness calibration, phylogenetic structure, codon-level mutation biases, indels, and the integration of experimental data.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript is a review summarizing how generative models (with emphasis on Direct Coupling Analysis) trained on homologous protein sequence families infer probabilistic landscapes that capture evolutionary constraints. These landscapes are then coupled to population-genetic or substitution-model dynamics to simulate protein evolution on experimental and phylogenetic timescales. The review covers landscape inference and validation, applications (viral evolution, laboratory drift, historical contingency, entrenchment, epistatic drift), and open challenges (score-fitness calibration, phylogenetic structure, codon biases, indels, experimental data integration). No new empirical results or derivations are presented; the central contribution is descriptive synthesis of existing literature.
Significance. If the coverage is accurate and balanced, the review offers a timely synthesis bridging generative sequence modeling and evolutionary dynamics, which could help researchers navigate connections between DCA-style approaches and population-genetic simulations. Explicitly flagging unresolved issues (rather than claiming resolution) is a strength. As a review without new results, its significance rests on the quality of literature representation and the utility of the outlined framework for future work.
minor comments (1)
- [Abstract] Abstract: the phrase 'we first describe how generative sequence landscapes are inferred and assessed, then review how they can be coupled...' would benefit from an explicit section-by-section outline early in the introduction to improve navigation for readers.
Simulated Author's Rebuttal
We thank the referee for their positive and accurate summary of the manuscript, which correctly identifies it as a review synthesizing existing literature on generative sequence models (with emphasis on DCA) for evolutionary dynamics. We appreciate the recommendation to accept and the recognition that explicitly flagging open challenges is a strength.
Circularity Check
No significant circularity in this review manuscript
full rationale
This manuscript is explicitly a review summarizing prior literature on generative sequence models (primarily DCA) and their coupling to population-genetic or substitution dynamics. It contains no new derivations, equations, fitted parameters, or quantitative predictions. All central claims are descriptive of approaches already explored in the cited external works; open challenges are flagged without resolution. No load-bearing step reduces by construction to inputs or self-citations.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
E. L. Van Dijk, H. Auger, Y. Jaszczyszyn, and C. Ther- mes, Ten years of next-generation sequencing technol- ogy, Trends in genetics30, 418 (2014)
2014
-
[2]
D. H. Ackley, G. E. Hinton, and T. J. Sejnowski, A learning algorithm for boltzmann machines, Cognitive science9, 147 (1985)
1985
-
[3]
Vaswani, N
A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser, and I. Polosukhin, At- tention is all you need, Advances in neural information processing systems30, 10. 48550/ arXiv. 1706. 03762 (2017)
2017
-
[4]
Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli,et al., Evolutionary-scale prediction of atomic-level protein structure with a language model, Science379, 1123 (2023)
2023
-
[5]
Stärk, C
H. Stärk, C. Dallago, M. Heinzinger, and B. Rost, Light attention predicts protein location from the language of life, Bioinformatics Advances1, vbab035 (2021)
2021
-
[6]
Cheng, C
P. Cheng, C. Mao, J. Tang, S. Yang, Y. Cheng, W. Wang, Q. Gu, W. Han, H. Chen, S. Li,et al., Zero- shot prediction of mutation effects with multimodal deep representation learning guides protein engineering, Cell Research34, 630 (2024)
2024
-
[7]
Madani, B
A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser, and N. Naik, Large language models generate functional protein sequences across diverse families, Nature Biotechnology41, 1099 (2023)
2023
-
[8]
Fragata, A
I. Fragata, A. Blanckaert, M. A. D. Louro, D. A. Liber- les, and C. Bank, Evolution in the light of fitness land- scape theory, Trends in ecology & evolution34, 69 (2019)
2019
-
[9]
J. F. Kingman, A simple model for the balance between selection and mutation, Journal of Applied Probability 15, 1 (1978)
1978
-
[10]
Kauffman and S
S. Kauffman and S. Levin, Towards a general theory of adaptive walks on rugged landscapes, Journal of theo- retical Biology128, 11 (1987)
1987
-
[11]
S. A. Kauffman and E. D. Weinberger, The nk model of rugged fitness landscapes and its application to mat- uration of the immune response, Journal of theoretical biology141, 211 (1989)
1989
-
[12]
R. A. Fisher,The genetical theory of natural selection: a complete variorum edition(Oxford University Press, 1999)
1999
-
[13]
Tenaillon, The utility of fisher’s geometric model in evolutionary genetics, Annual review of ecology, evolu- tion, and systematics45, 179 (2014)
O. Tenaillon, The utility of fisher’s geometric model in evolutionary genetics, Annual review of ecology, evolu- tion, and systematics45, 179 (2014)
2014
-
[14]
Pahujani and J
S. Pahujani and J. Krug, Complexity and accessibility of random landscapes, SciPost Phys. Lect. Notes , 108 (2025)
2025
-
[15]
Evolution as fitness landscape navigation: Concepts, Measures, and Emerging Questions
M. Srivastava, C. Bank, J. Krug, and S. G. Das, Evo- lution as fitness landscape navigation: Concepts, mea- sures, and emerging questions, arXiv 2604.17036 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[16]
Simple sign epistasis and evolutionary detours in fitness landscapes
P. Ribeca, A. Castro, A. Lage-Castellanos, A. Sergeeva, S. Matuszewski, R. G. Paccosi, V. Belik, M. Ghafari, J. Krug, G. Achaz, and L. Ferretti, Simple sign epistasis and evolutionary detours in fitness landscapes, arXiv 2604.22611 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[17]
D. M. Fowler and S. Fields, Deep mutational scanning: a new style of protein science, Nature methods11, 801 (2014)
2014
-
[18]
Firnberg, J
E. Firnberg, J. W. Labonte, J. J. Gray, and M. Oster- meier, A comprehensive, high-resolution map of a gene’s fitness landscape, Molecular biology and evolution31, 1581 (2014)
2014
-
[19]
F. J. Poelwijk, M. Socolich, and R. Ranganathan, Learning the pattern of epistasis linking genotype and phenotype in a protein, Nature communications10, 4213 (2019)
2019
-
[20]
Papkou, L
A. Papkou, L. Garcia-Pastor, J. A. Escudero, and A. Wagner, A rugged yet easily navigable fitness land- scape, Science382, eadh3860 (2023)
2023
-
[21]
K. Buda, C. M. Miton, and N. Tokuriki, Pervasive epistasis exposes intramolecular networks in adaptive enzyme evolution, Nature Communications14, 8508 (2023)
2023
-
[22]
J. A. G. De Visser and J. Krug, Empirical fitness land- scapes and the predictability of evolution, Nature Re- views Genetics15, 480 (2014)
2014
-
[23]
T. N. Starr and J. W. Thornton, Epistasis in protein evolution, Protein science25, 1204 (2016)
2016
-
[24]
M. S. Johnson, G. Reddy, and M. M. Desai, Epistasis and evolution: recent advances and an outlook for pre- diction, BMC biology21, 120 (2023)
2023
-
[25]
C. M. Miton and N. Tokuriki, How mutational epistasis impairs predictability in protein evolution and design, Protein Science25, 1260 (2016)
2016
-
[26]
M. Blum, A. Andreeva, L. C. Florentino, S. R. Chugu- ransky, T. Grego, E. Hobbs, B. L. Pinto, A. Orr, T. Paysan-Lafosse, I. Ponamareva,et al., Interpro: the protein sequence classification resource in 2025, Nucleic Acids Research , gkae1082 (2024)
2025
-
[27]
D. A. Liberles, S. A. Teichmann, I. Bahar, U. Bas- tolla, J. Bloom, E. Bornberg-Bauer, L. J. Colwell, A. J. De Koning, N. V. Dokholyan, J. Echave,et al., The interface of protein structure, protein biophysics, and molecular evolution, Protein Science21, 769 (2012)
2012
-
[28]
T. Sikosek and H. S. Chan, Biophysics of protein evo- lution and evolutionary protein biophysics, Journal of The Royal Society Interface11, 10.1098/rsif.2014.0419 (2014)
-
[29]
Bastolla, Y
U. Bastolla, Y. Dehouck, and J. Echave, What evolu- tion tells us about protein physics, and protein physics tells us about evolution, Current opinion in structural biology42, 59 (2017)
2017
-
[30]
Echave and C
J. Echave and C. O. Wilke, Biophysical models of pro- tein evolution: understanding the patterns of evolution- ary sequence divergence, Annual review of biophysics 46, 85 (2017)
2017
-
[31]
A. S. Lapedes, B. G. Giraud, L. Liu, and G. D. Stormo, Correlated mutations in models of protein se- quences: phylogenetic and structural effects, Lecture Notes-Monograph Series , 236 (1999)
1999
-
[32]
Weigt, R
M. Weigt, R. A. White, H. Szurmant, J. A. Hoch, and T. Hwa, Identification of direct residue contacts in protein–protein interaction by message passing, Pro- ceedings of the National Academy of Sciences106, 67 (2009). 23
2009
-
[33]
Bjerregaard, P
A. Bjerregaard, P. M. Groth, S. Hauberg, A. Krogh, and W. Boomsma, Foundation models of protein sequences: A brief overview, Current Opinion in Structural Biology 91, 103004 (2025)
2025
-
[34]
Figliuzzi, P
M. Figliuzzi, P. Barrat-Charlaix, and M. Weigt, How pairwise coevolutionary models capture the collective residue variability in proteins?, Molecular biology and evolution35, 1018 (2018)
2018
-
[35]
McGee, S
F. McGee, S. Hauri, Q. Novinger, S. Vucetic, R. M. Levy, V. Carnevale, and A. Haldane, The generative ca- pacity of probabilistic protein sequence models, Nature communications12, 6302 (2021)
2021
-
[36]
Shekhar, C
K. Shekhar, C. F. Ruberman, A. L. Ferguson, J. P. Bar- ton, M. Kardar, and A. K. Chakraborty, Spin models inferred from patient-derived viral sequence data faith- fully describe hiv fitness landscapes, Physical Review E—Statistical, Nonlinear, and Soft Matter Physics88, 062705 (2013)
2013
-
[37]
Kaltenbach and N
M. Kaltenbach and N. Tokuriki, Dynamics and con- straints of enzyme evolution, Journal of Experimental Zoology Part B: Molecular and Developmental Evolu- tion322, 468 (2014)
2014
-
[38]
Morcos, T
F. Morcos, T. Hwa, J. N. Onuchic, and M. Weigt, Di- rect coupling analysis for protein contact prediction, in Protein structure prediction(Springer, 2014) pp. 55–70
2014
-
[39]
R. M. Levy, A. Haldane, and W. F. Flynn, Potts hamil- tonian models of protein co-variation, free energy land- scapes, and evolutionary fitness, Current opinion in structural biology43, 55 (2017)
2017
-
[40]
Cocco, C
S. Cocco, C. Feinauer, M. Figliuzzi, R. Monasson, and M. Weigt, Inverse statistical physics of protein se- quences: a key issues review, Reports on Progress in Physics81, 032601 (2018)
2018
-
[41]
Haldane and R
A. Haldane and R. M. Levy, Influence of multiple- sequence-alignment depth on potts statistical models of protein covariation, Physical Review E99, 032405 (2019)
2019
-
[42]
Tiana and R
G. Tiana and R. Broglia, The molecular evolution of HIV-1 protease simulated at atomic detail, Proteins: Structure, Function, and Bioinformatics76, 895 (2009)
2009
-
[43]
Crippa, D
M. Crippa, D. Andreghetti, R. Capelli, and G. Tiana, Evolutionoffrustratedandstabilisingcontactsinrecon- structed ancient proteins, European Biophysics Journal 50, 699 (2021)
2021
-
[44]
Socolich, S
M. Socolich, S. W. Lockless, W. P. Russ, H. Lee, K. H. Gardner, and R. Ranganathan, Evolutionary informa- tion for specifying a protein fold, Nature437, 512 (2005)
2005
-
[45]
Bisardi, J
M. Bisardi, J. Rodriguez-Rivas, F. Zamponi, and M. Weigt, Modeling sequence-space exploration and emergence of epistatic signals in protein evolution, Molecular biology and evolution39, msab321 (2022)
2022
-
[46]
Lunzer, G
M. Lunzer, G. B. Golding, and A. M. Dean, Pervasive cryptic epistasis in molecular evolution, PLoS genetics 6, e1001162 (2010)
2010
-
[47]
Biswas, A
A. Biswas, A. Haldane, E. Arnold, and R. M. Levy, Epistasis and entrenchment of drug resistance in hiv-1 subtype b, Elife8, e50524 (2019)
2019
-
[48]
Domingo, P
J. Domingo, P. Baeza-Centurion, and B. Lehner, The causes and consequences of genetic interactions (epista- sis), Annual review of genomics and human genetics20, 433 (2019)
2019
-
[49]
Y. Park, B. P. Metzger, and J. W. Thornton, Epistatic drift causes gradual decay of predictability in protein evolution, Science376, 823 (2022)
2022
-
[50]
J. Chen, M. Bisardi, D. Lee, S. Cotogno, F. Zamponi, M.Weigt,andN.Tokuriki,Understandingepistaticnet- works in the b1β-lactamases through coevolutionary statistical modeling and deep mutational scanning, Na- ture communications15, 8441 (2024)
2024
-
[51]
Ekeberg, C
M. Ekeberg, C. Lövkvist, Y. Lan, M. Weigt, and E. Au- rell, Improved contact prediction in proteins: using pseudolikelihoods to infer potts models, Physical Re- view E—Statistical, Nonlinear, and Soft Matter Physics 87, 012707 (2013)
2013
-
[52]
Trinquier, G
J. Trinquier, G. Uguzzoni, A. Pagnani, F. Zamponi, and M. Weigt, Efficient generative modeling of protein sequences using simple autoregressive models, Nature communications12, 5800 (2021)
2021
-
[53]
Morcos, A
F. Morcos, A. Pagnani, B. Lunt, A. Bertolino, D. S. Marks, C. Sander, R. Zecchina, J. N. Onuchic, T. Hwa, and M. Weigt, Direct-coupling analysis of residue co- evolution captures native contacts across many protein families, Proceedings of the National Academy of Sci- ences108, E1293 (2011)
2011
-
[54]
Cocco and R
S. Cocco and R. Monasson, Adaptive cluster expansion for inferring boltzmann machines with noisy data, Phys- ical review letters106, 090601 (2011)
2011
-
[55]
Barrat-Charlaix, A
P. Barrat-Charlaix, A. P. Muntoni, K. Shimagaki, M. Weigt, and F. Zamponi, Sparse generative modeling via parameter reduction of boltzmann machines: appli- cation to protein-sequence families, Physical Review E 104, 024407 (2021)
2021
-
[56]
Tsishyn, H
M. Tsishyn, H. Talibart, M. Rooman, and F. Pucci, Structure-informed direct coupling analysis improves protein mutational landscape predictions, bioRxiv , 2026 (2026)
2026
-
[57]
Haldane and R
A. Haldane and R. M. Levy, Mi3-gpu: Mcmc-based inverse ising inference on gpus for protein covaria- tion analysis, Computer physics communications260, 107312 (2021)
2021
-
[58]
Rosset, R
L. Rosset, R. Netti, A. P. Muntoni, M. Weigt, and F. Zamponi, adabmDCA 2.0—a flexible but easy-to-use package for direct coupling analysis, inProtein Evolu- tion: Methods and Protocols, Methods in Molecular Bi- ology, Vol. 2979, edited by S. M. Khan and F. Pazos (Humana, New York, NY, 2026) pp. 83–104
2026
-
[59]
W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat- Charlaix, M.Socolich, P.Kast, D.Hilvert, R.Monasson, S. Cocco, M. Weigt,et al., An evolution-based model for designing chorismate mutase enzymes, Science369, 440 (2020)
2020
-
[60]
B. Fram, Y. Su, I. Truebridge, A. J. Riesselman, J. B. Ingraham, A. Passera, E. Napier, N. N. Thadani, S. Lim, K. Roberts,et al., Simultaneous enhancement of multiple functional properties using evolution-informed protein design, Nature communications15, 5141 (2024)
2024
-
[61]
Fernandez-de Cossio-Diaz, P
J. Fernandez-de Cossio-Diaz, P. Hardouin, F.-X. Ly- onnet du Moutier, A. Di Gioacchino, B. Marchand, Y. Ponty, B. Sargueil, R. Monasson, and S. Cocco, De- signing molecular rna switches with restricted boltz- mann machines, Nature Communications16, 11223 (2025)
2025
-
[62]
Expanding functional protein sequence space using high entropy generative models
R. Netti, E. Hinds, F. Calvanese, R. Ranganathan, M. Weigt, and F. Zamponi, Expanding functional pro- tein sequence space using high entropy generative mod- els, arXiv 2605.03578 (2026). 24
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[63]
Romero-Romero, S
S. Romero-Romero, S. Lindner, and N. Ferruz, Explor- ing the protein sequence space with global generative models, Cold Spring Harbor Perspectives in Biology15, a041471 (2023)
2023
-
[64]
Mardikoraem, Z
M. Mardikoraem, Z. Wang, N. Pascual, and D. Woldring, Generative models for protein sequence modeling: recent advances and future directions, Briefings in Bioinformatics24, bbad358 (2023)
2023
-
[65]
C. Hsu, H. Nisonoff, C. Fannjiang, and J. Listgarten, Learning protein fitness models from evolutionary and assay-labeled data, Nature biotechnology40, 1114 (2022)
2022
-
[66]
Tubiana, S
J. Tubiana, S. Cocco, and R. Monasson, Learning pro- tein constitutive motifs from sequence data, Elife8, e39397 (2019)
2019
-
[67]
Shimagaki and M
K. Shimagaki and M. Weigt, Selection of sequence mo- tifs and generative hopfield-potts models for protein families, Physical Review E100(2019)
2019
-
[68]
Decelle, A
A. Decelle, A. d. J. Navas Gómez, and B. Seoane, Infer- ring higher-order couplings with neural networks, Phys. Rev. Lett.135, 207301 (2025)
2025
-
[69]
M. Huot, P. Rosenbaum, C. Planchais, H. Mouquet, R. Monasson, and S. Cocco, Generative model of sars-cov-2 variants under immune pressure unveils vi- ralescapepotential,bioRxiv10.1101/2025.05.12.653592 (2025)
2025
-
[70]
M. Huot, D. Wang, E. Shakhnovich, R. Monasson, and S. Cocco, Constrained evolutionary funnels shape viral immune escape, Proceedings of the National Academy of Sciences123, e2536956123 (2026)
2026
-
[71]
A. Rehan, E. Mauri, J. Fernandez-de Cossio-Diaz, P.-G. Brun, R. Monasson, M. Ribezzi-Crivellari, and S. Cocco, Design and experimental characterization of specificity-switching mutational paths of ww domains, bioRxiv 10.64898/2025.12.08.693000 (2025)
-
[72]
X. Ding, Z. Zou, and C. L. Brooks, Deciphering pro- tein evolution and fitness landscapes with latent space models, Nature Communications10, 10.1038/s41467- 019-13633-0 (2019)
-
[73]
Hawkins-Hooker, F
A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Coua- iron, A. Chen, and D. Bikard, Generating functional protein variants with variational autoencoders, PLoS computational biology17, e1008736 (2021)
2021
-
[74]
C. Ziegler, J. Martin, C. Sinner, and F. Morcos, Latent generative landscapes as maps of functional diversity in protein sequence space, Nature Communications14, 10.1038/s41467-023-37958-z (2023)
-
[75]
D. Shukla, J. Martin, F. Morcos, and D. A. Potoyan, Thermal adaptation of cytosolic malate dehydrogenase revealed by deep learning and coevolutionary anal- ysis, Journal of Chemical Theory and Computation 10.1021/acs.jctc.4c01774 (2025)
-
[76]
Rives, J
A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin, J. Liu, D. Guo, M. Ott, C. L. Zitnick, J. Ma,et al., Bio- logical structure and function emerge from scaling un- supervised learning to 250 million protein sequences, Proceedings of the National Academy of Sciences118, e2016239118 (2021)
2021
-
[77]
D. Hesslow, N. Zanichelli, P. Notin, I. Poli, and D.Marks,Rita: astudyonscalingupgenerativeprotein sequence models, arXiv 2205.05789 (2022)
-
[78]
Sevgen, J
E. Sevgen, J. Moller, A. Lange, J. Parker, S. Quigley, J. Mayer, P. Srivastava, S. Gayatri, D. Hosfield, C. Dilks,et al., Prot-vae: protein transformer varia- tional autoencoder for functional protein design, Pro- ceedings of the National Academy of Sciences122, e2408737122 (2025)
2025
-
[79]
Notin, M
P. Notin, M. Dias, J. Frazer, J. Marchena-Hurtado, A. N. Gomez, D. Marks, and Y. Gal, Tranception: pro- tein fitness prediction with autoregressive transformers and inference-time retrieval, inInternational Confer- ence on Machine Learning(PMLR, 2022) pp. 16990– 17017
2022
-
[80]
A.Chandra, L.Tünnermann, T.Löfstedt,andR.Gratz, Transformer-based deep learning for predicting protein properties in the life sciences, Elife12, e82819 (2023)
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.