A Large-Scale Dataset for Molecular Structure-Language Description via a Rule-Regularized Method
Pith reviewed 2026-05-16 08:08 UTC · model grok-4.3
The pith
An automated framework parses IUPAC names into structural metadata to guide LLMs in creating a 163000-pair molecule-description dataset at 98.6 percent precision.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
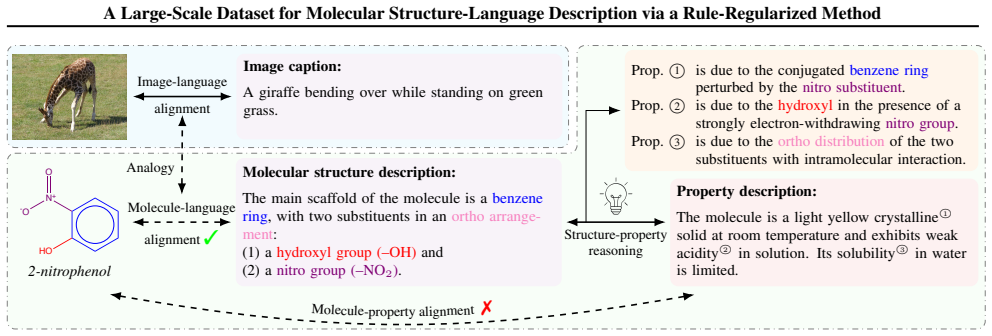
By extending a rule-based chemical nomenclature parser to produce enriched structural XML metadata from IUPAC names and then using that metadata to constrain LLM generation, the work creates a scalable, high-precision collection of approximately 163000 molecule-description pairs that preserve complete structural details.
What carries the argument
The rule-regularized annotation framework that converts IUPAC names into structural XML metadata to guide subsequent LLM description generation.
If this is right
- Models trained on the dataset can perform chemical reasoning tasks that require accurate structure-language alignment.
- The automated pipeline supplies a reusable template for creating similar grounded datasets in other domains that use structured scientific nomenclature.
- Chemical applications that depend on precise structural descriptions, such as property prediction or reaction planning, gain access to a larger training resource than previously available.
- Validation methods that combine automated LLM checks with targeted human review become a practical standard for maintaining quality in large-scale scientific annotation efforts.
Where Pith is reading between the lines
- The same parser-plus-LLM pattern could be tested on other structured naming systems, such as protein sequences or crystal notations, to produce cross-domain description datasets.
- Performance gains on downstream chemical benchmarks could be measured by fine-tuning models on this dataset versus existing smaller or noisier collections.
- Further rule extensions might allow the framework to handle molecules whose IUPAC names currently fall outside the parser's coverage, increasing dataset breadth.
Load-bearing premise
The extended rule-based parser extracts every structural detail correctly from any IUPAC name into XML metadata, and the LLMs then generate descriptions that match that metadata without adding or omitting structural facts.
What would settle it
Independent expert review of a large random sample of the generated descriptions, checked directly against the source molecular graphs, would reveal frequent structural mismatches or omissions beyond the reported 98.6 percent precision on the 2000-molecule validation set.
Figures

read the original abstract
Molecular function is largely determined by structure. Accurately aligning molecular structure with natural language is therefore essential for enabling large language models (LLMs) to reason about downstream chemical tasks. However, the substantial cost of human annotation makes it infeasible to construct large-scale, high-quality datasets of structure-grounded descriptions. In this work, we propose a fully automated annotation framework for generating precise molecular descriptions that preserve complete structural details at scale. Our approach builds upon and extends a rule-based chemical nomenclature parser to interpret IUPAC names and construct enriched, structural XML metadata that explicitly encodes molecular structure. This metadata is then used to guide LLMs in producing accurate natural-language descriptions. Using this framework, we curate a large-scale dataset of approximately $163$k molecule--description pairs. A rigorous validation protocol combining LLM-based and expert human evaluation on a subset of $2,000$ molecules demonstrates a high description precision of $98.6$%. The proposed annotation framework is readily beneficial to broader chemical tasks that rely on structural descriptions, with the resulting dataset providing a reliable foundation for molecule--language alignment. The source code and dataset are hosted at https://github.com/TheLuoFengLab/MolLangData and https://huggingface.co/datasets/ChemFM/MolLangData, respectively.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes a fully automated framework that extends a rule-based chemical nomenclature parser to convert IUPAC names into enriched structural XML metadata, which then guides LLMs to generate natural-language molecular descriptions. Using this pipeline the authors curate a dataset of ~163k molecule–description pairs and report 98.6% description precision on a 2,000-molecule validation subset evaluated by both LLM-based and expert human judges.
Significance. A reliably constructed, large-scale, structure-grounded molecule–language dataset would be a valuable resource for training and evaluating LLMs on chemical reasoning tasks. The reported scale and the public release of code and data are concrete strengths; however, the significance is conditional on the parser’s coverage and the representativeness of the validation subset.
major comments (2)
- [§3] §3: The extended rule-based parser is presented without any quantitative coverage statistics, failure-mode enumeration, or error-rate measurement over the full PubChem-derived IUPAC name distribution. Because the central claim of 98.6% precision on the entire 163k set rests on every IUPAC name yielding complete structural XML, the absence of such analysis is load-bearing.
- [abstract and §4] Validation protocol (abstract and §4): No information is given on how the 2,000-molecule subset was sampled, whether it was stratified by structural complexity, or what exact criteria defined a “precise” description. This leaves open the possibility that the reported precision does not generalize to the full dataset.
minor comments (2)
- The abstract states that the framework is “readily beneficial to broader chemical tasks,” but the manuscript provides no concrete downstream experiments or transfer results to support this claim.
- Notation for the XML schema and the precise mapping from parser output fields to LLM prompt tokens is not fully specified, making reproducibility of the generation step difficult.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. We address each of the major comments below and have revised the manuscript accordingly to strengthen the presentation of our methods and validation.
read point-by-point responses
-
Referee: [§3] §3: The extended rule-based parser is presented without any quantitative coverage statistics, failure-mode enumeration, or error-rate measurement over the full PubChem-derived IUPAC name distribution. Because the central claim of 98.6% precision on the entire 163k set rests on every IUPAC name yielding complete structural XML, the absence of such analysis is load-bearing.
Authors: We acknowledge that the original manuscript lacked quantitative coverage statistics for the extended rule-based parser. This is a valid point, and we have added a detailed analysis in the revised Section 3, including coverage rates, failure modes, and error rates on the PubChem-derived distribution. This supports the claim that the pipeline produces complete structural XML for the curated dataset. revision: yes
-
Referee: [abstract and §4] Validation protocol (abstract and §4): No information is given on how the 2,000-molecule subset was sampled, whether it was stratified by structural complexity, or what exact criteria defined a “precise” description. This leaves open the possibility that the reported precision does not generalize to the full dataset.
Authors: We thank the referee for highlighting the need for more details on the validation protocol. In the revised manuscript, we have clarified in the abstract and Section 4 that the 2,000-molecule subset was randomly sampled from the 163k dataset. We have also specified the criteria for precision (accurate inclusion of all structural features without errors or omissions) and added evidence that the subset is representative in terms of structural complexity. revision: yes
Circularity Check
No circularity in dataset generation claims
full rationale
The paper describes an automated pipeline that extends a rule-based IUPAC parser to produce structural XML metadata, then uses that metadata to prompt LLMs for natural-language descriptions, resulting in a 163k-pair dataset whose quality is measured by separate LLM-based and human evaluation on a 2,000-sample subset (98.6% precision). No equations, fitted parameters, or predictions appear in the provided text. The central claim rests on external validation rather than any self-referential derivation, self-citation chain, or renaming of inputs. The method is therefore self-contained with independent quality assessment and exhibits no circularity.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard IUPAC nomenclature rules can be parsed to accurately represent molecular structures in XML metadata.
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.