Towards coevolution-aware ancestral sequence reconstruction
Pith reviewed 2026-06-29 02:08 UTC · model grok-4.3
The pith
A framework combines phylogenetic inference with direct coupling analysis to reconstruct ancestral proteins under epistatic constraints.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
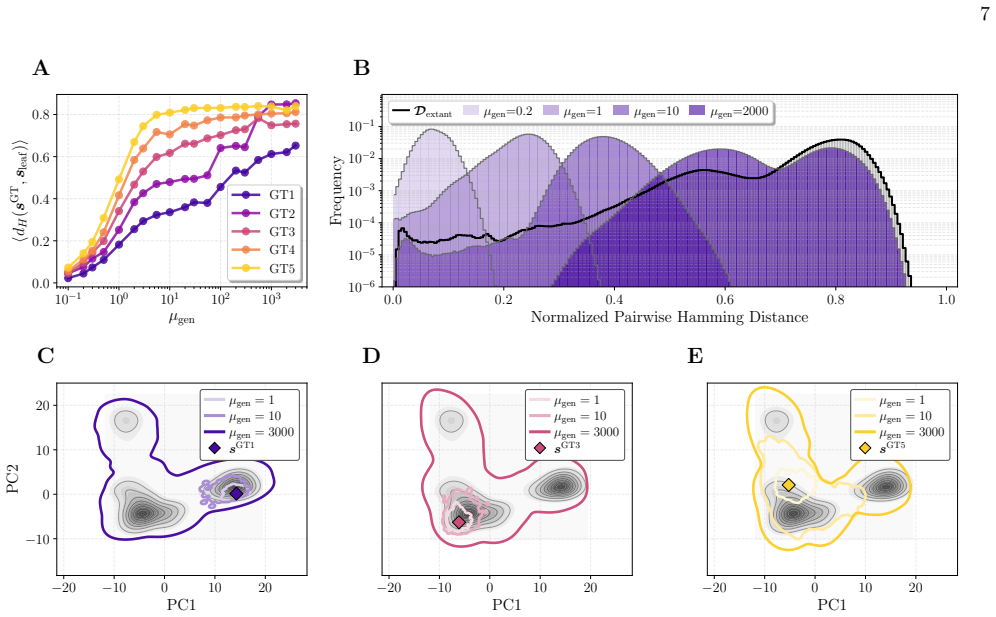
The coevolution-aware ASR framework integrates standard phylogenetic inference with Direct Coupling Analysis learned from extant protein families. It preserves site-wise ancestral uncertainty while enforcing the learned residue-residue constraints. When tested against known ground-truth sequences generated by a DCA-based forward-evolution sampler on beta-lactamases and DNA-binding domains, the method improves reconstruction accuracy under epistatic constraints and yields ensembles that remain both phylogenetically consistent and statistically compatible with natural protein families.
What carries the argument
The coevolution-aware ASR framework that merges phylogenetic inference with DCA to enforce epistatic constraints while retaining uncertainty.
If this is right
- Reconstructed ancestors match ground-truth sequences more closely when epistatic constraints are present.
- Generated ensembles remain phylogenetically consistent with the input tree.
- Ensembles are statistically compatible with the sequence statistics of natural protein families.
- The approach supplies an intermediate between single MAP sequences and fully unconstrained posterior samples.
Where Pith is reading between the lines
- The method could be tested on additional protein families to check whether the improvement scales with the strength of observed epistasis.
- It opens the possibility of using the generated ensembles to predict which ancient functions would have been compatible with coevolutionary patterns.
- Extensions might replace DCA with other coupling models to assess robustness across different representations of epistasis.
Load-bearing premise
Coevolutionary constraints inferred by DCA from extant sequences remain valid and representative for the ancestral sequences being reconstructed.
What would settle it
If the new reconstructions show no higher match to ground-truth sequences than independent-site methods when tested on DCA-simulated forward-evolution trajectories with strong epistasis, the claimed improvement does not hold.
Figures




read the original abstract
Ancestral sequence reconstruction (ASR) is a powerful approach for studying molecular evolution and the emergence of protein function. Yet most ASR methods assume that sites evolve independently, neglecting the epistatic constraints that shape protein structure, stability, and function. This simplification affects both ancestral inference and its evaluation: maximum-a-posteriori reconstructions may over-concentrate probability into a single over-idealized sequence, whereas independent posterior sampling can generate implausible or poorly functional ancestors. Here, we introduce a coevolution-aware ASR framework that combines standard phylogenetic inference with Direct Coupling Analysis (DCA), thereby preserving site-wise ancestral uncertainty while enforcing residue-residue constraints learned from extant protein families. To benchmark the method, we develop a controlled forward-evolution framework based on a DCA evolutionary sampler, allowing reconstructed ancestors to be compared with known ground-truth sequences generated under realistic epistatic constraints. Applied to beta-lactamases and DNA-binding domains, the approach improves reconstruction when ancestral states are epistatically constrained, and yields ensembles of candidate ancestors that are both phylogenetically consistent and statistically compatible with natural protein families. This framework bridges the gap between single-sequence MAP reconstruction and unconstrained posterior sampling, providing a practical route toward ancestral reconstructions that better reflect the coupled nature of protein evolution.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a coevolution-aware ancestral sequence reconstruction (ASR) framework that augments standard phylogenetic inference with Direct Coupling Analysis (DCA) to enforce residue-residue epistatic constraints learned from extant sequences. It develops a controlled forward-evolution benchmark that generates ground-truth ancestral sequences via a DCA-based evolutionary sampler, enabling direct comparison of reconstructions against known sequences under the modeling assumptions. The method is applied to beta-lactamase and DNA-binding domain families, with the central claim that it improves reconstruction accuracy when ancestral states are epistatically constrained and produces ensembles that remain both phylogenetically consistent and statistically compatible with natural protein families.
Significance. If the central claims hold after addressing validation gaps, the work would provide a practical method for generating more realistic ancestral sequence ensembles that respect coevolutionary structure, addressing a known limitation of independent-site ASR models. The controlled forward-evolution benchmark is a clear strength, as it supplies a reproducible testbed under known epistatic constraints rather than relying solely on real-data proxies.
major comments (2)
- [Abstract] Abstract: The claim that the approach 'improves reconstruction when ancestral states are epistatically constrained' on the two real families is stated without any quantitative metrics (e.g., per-site recovery rates, Hamming distance to ground truth, or statistical significance relative to independent-site baselines). This absence is load-bearing because the central claim of practical improvement cannot be assessed from the supplied text.
- [Benchmark description (Methods/Results)] Benchmark description (Methods/Results): The forward-evolution framework generates ground-truth sequences from the identical DCA model subsequently used for reconstruction and constraint enforcement. While this establishes internal consistency under the modeling assumptions, it provides no test of whether DCA couplings inferred from extant sequences remain representative for deep ancestral states in real proteins (e.g., via contact gain/loss). This assumption is load-bearing for the claims on beta-lactamases and DNA-binding domains.
minor comments (1)
- [Methods] The description of how phylogenetic posteriors are combined with DCA constraints during sampling would benefit from an explicit algorithmic outline or pseudocode to clarify the procedure for readers.
Simulated Author's Rebuttal
We thank the referee for the thoughtful and constructive report. The comments highlight important aspects of how claims are presented and the scope of the benchmark validation. We address each point below and will revise the manuscript to improve clarity and transparency.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that the approach 'improves reconstruction when ancestral states are epistatically constrained' on the two real families is stated without any quantitative metrics (e.g., per-site recovery rates, Hamming distance to ground truth, or statistical significance relative to independent-site baselines). This absence is load-bearing because the central claim of practical improvement cannot be assessed from the supplied text.
Authors: We agree that the abstract should be self-contained. The quantitative support for the improvement claim (per-site recovery rates, Hamming distances to ground-truth ancestors, and statistical comparisons against independent-site baselines) is provided in the Results section for the forward-evolution benchmarks performed on the beta-lactamase and DNA-binding domain families. We will revise the abstract to incorporate the key numerical results and significance statements so that the central claim can be evaluated directly from the abstract. revision: yes
-
Referee: [Benchmark description (Methods/Results)] Benchmark description (Methods/Results): The forward-evolution framework generates ground-truth sequences from the identical DCA model subsequently used for reconstruction and constraint enforcement. While this establishes internal consistency under the modeling assumptions, it provides no test of whether DCA couplings inferred from extant sequences remain representative for deep ancestral states in real proteins (e.g., via contact gain/loss). This assumption is load-bearing for the claims on beta-lactamases and DNA-binding domains.
Authors: We acknowledge that the benchmark tests recovery under the assumption that the DCA model inferred from extant sequences remains valid for ancestral states. This is a standard modeling choice for controlled simulation but does not directly probe whether couplings or contacts evolve over deep time. The simulation nevertheless isolates the benefit of epistatic constraints relative to independent-site models. For the real-family applications we further show that the output ensembles remain statistically compatible with the natural sequence distribution and phylogenetically consistent. We will add an explicit discussion of this benchmark limitation and its implications in the revised manuscript. revision: partial
Circularity Check
No significant circularity; derivation combines independent standard components
full rationale
The paper's central framework integrates standard phylogenetic inference (independent of the present work) with DCA couplings inferred from extant sequences, then applies those couplings during ancestral sampling. The forward-evolution benchmark generates ground-truth sequences from a DCA model and recovers them with the same model; this is a conventional internal-consistency test under known generative assumptions, not a reduction of the claimed improvement to a fitted parameter or self-definition. Real-family results rest on the external (and separately debatable) assumption that DCA constraints are time-invariant, but that assumption is not smuggled in via self-citation, ansatz, or renaming; the derivation chain therefore remains self-contained against external benchmarks and does not match any enumerated circularity pattern.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption DCA models trained on extant sequences capture the epistatic constraints that governed ancestral sequences
- domain assumption Standard phylogenetic substitution models remain appropriate once site couplings are added via DCA
Reference graph
Works this paper leans on
-
[1]
We first perform standard site- independent ASR using the Yang et al
Site-independent reconstruction (Section Meth- ods IV E). We first perform standard site- independent ASR using the Yang et al. algo- rithm [7] to infer the per-site posterior distribu- tion of amino acids at the root node. Rather than using a traditional substitution model (WAG [22], LG [49], or JTT [50]), or even more complex ones that incorporate heuri...
-
[2]
We drawM= 1000 candidate root sequences from the site- independent posteriorP R(· |D,T,θ), obtaining the candidate setS anc
Sampling of candidate ancestors. We drawM= 1000 candidate root sequences from the site- independent posteriorP R(· |D,T,θ), obtaining the candidate setS anc. 5
-
[3]
A siteiand two sequencess α,s β ∈S anc are cho- sen at random, and their amino acids at siteiare swapped to obtain sequences ˆsα and ˆsβ
Coevolutionary reshuffling (Section Methods IV F). A siteiand two sequencess α,s β ∈S anc are cho- sen at random, and their amino acids at siteiare swapped to obtain sequences ˆsα and ˆsβ. The swap is accepted with probability min 1, e−∆Ei,αβ DCA/T , where ∆Ei,αβ DCA = EDCA(ˆsα) +E DCA(ˆsβ) − EDCA(sα) +E DCA(sβ) , (4) andTis an effective temperature contr...
2000
-
[4]
Independent Site Reconstruction: We first perform the site-independent ancestral reconstruction to ob- tain the site-wise posterior distributionsP (i) R at the root of the tree
-
[5]
This yields a site-independent alignment of candidate ancestors Sanc = (s α R,i)α∈{1,···,M} i∈{1,···,L} , where columns represent sites and rows represent individual sequences
Initial Sampling: A set ofM= 1000 candidate an- cestral sequences is sampled from the product pos- terior distributionP R = QL i=1 P(i) R . This yields a site-independent alignment of candidate ancestors Sanc = (s α R,i)α∈{1,···,M} i∈{1,···,L} , where columns represent sites and rows represent individual sequences. The choice ofMis a heuristic balance, pr...
-
[6]
Coevolutionary Optimization: The alignment is re- fined through a column-wise amino acid swap pro- cedure to minimize the DCA score. In each itera- tion: (a) A siteiis selected with probability propor- tional to its Shannon entropy Γ i (calculated from the frequencies inS anc), ensuring the op- timization focuses on non-conserved positions. (b) Two sequen...
-
[7]
Termination: The procedure stops heuristically af- ter 2M·Γ/2 attempted moves, where Γ =PL i=1 Γi is the total entropy of the alignment. For sitei, we define Γi =− P a∈A fi(a) log(fi(a)) as the site-wise entropy of the amino acid distribution of the an- cestral setS anc, taking the single-point frequencies fi of the amino acids as a probability distributi...
2023
-
[8]
restoration stud- ies
L. Pauling, E. Zuckerkandl, T. Henriksen, and R. L¨ ovs- tad, Chemical paleogenetics. molecular “restoration stud- ies” of extinct forms of life, Acta Chemica Scandinavica 17, 9 (1963)
1963
-
[9]
D. A. Liberles,Ancestral sequence reconstruction(OUP Oxford, 2007)
2007
-
[10]
M. J. Harms and J. W. Thornton, Analyzing pro- tein structure and function using ancestral gene recon- struction, Current opinion in structural biology20, 360 (2010)
2010
-
[11]
J. W. Thornton, Resurrecting ancient genes: experimen- tal analysis of extinct molecules, Nature Reviews Genet- ics5, 366 (2004)
2004
-
[12]
Prakinee, S
K. Prakinee, S. Phaisan, S. Kongjaroon, and P. Chaiyen, Ancestral Sequence Reconstruction for Designing Bio- catalysts and Investigating their Functional Mechanisms, JACS Au4, 4571 (2024)
2024
-
[13]
P. M. Zakas, H. C. Brown, K. Knight, S. L. Meeks, H. T. Spencer, E. A. Gaucher, and C. B. Doering, Enhancing the pharmaceutical properties of protein drugs by ances- tral sequence reconstruction, Nature biotechnology35, 35 (2017)
2017
-
[14]
Z. Yang, S. Kumar, and M. Nei, A New Method of Infer- ence of Ancestral Nucleotide and Amino Acid Sequences, Genetics141, 1641 (1995)
1995
-
[15]
Felsenstein, Evolutionary trees from DNA sequences: A maximum likelihood approach, Journal of Molecular Evolution17, 368 (1981)
J. Felsenstein, Evolutionary trees from DNA sequences: A maximum likelihood approach, Journal of Molecular Evolution17, 368 (1981)
1981
-
[16]
Cocco, C
S. Cocco, C. Feinauer, M. Figliuzzi, R. Monasson, and M. Weigt, Inverse statistical physics of protein sequences: a key issues review, Reports on Progress in Physics81, 032601 (2018)
2018
-
[17]
J. Zhao, B. Wang, J. Di, J. Zhou, J. Dong, Y. Ni, and R. Han, Ancestral Sequence Reconstruction for Novel Bi- functional Glutathione Synthetase with Enhanced Ther- mostability and Catalytic Efficiency, Foods15, 309 (2026)
2026
-
[18]
Chernyavskaya, M
E. Chernyavskaya, M. Vorobeva, S. A. Spirin, D. A. Skvortsov, and D. Pervouchine, Ancestral intronic splic- ing regulatory elements in the SCNαgene family, RNA , rna.080730.125 (2026)
2026
-
[19]
S. Supekar, W. L. Yeo, E. Tiong, J. Rizal, E. L. Ang, F. T. Wong, Y. H. Lim, and H. Fan, Ancestral Se- quence Reconstruction Reveals New Functional Fluori- nases and Mechanistic Insights into Enzymatic Fluorina- tion, Chemical Communications 10.1039/D5CC06378G (2026)
-
[20]
M. A. Spence, J. A. Kaczmarski, J. W. Saunders, and C. J. Jackson, Ancestral sequence reconstruction for pro- tein engineers, Current opinion in structural biology69, 131 (2021)
2021
-
[21]
G. N. Eick, J. T. Bridgham, D. P. Anderson, M. J. Harms, and J. W. Thornton, Robustness of Recon- structed Ancestral Protein Functions to Statistical Un- certainty, Molecular Biology and Evolution34, 247 (2017)
2017
-
[22]
P. D. Williams, D. D. Pollock, B. P. Blackburne, and R. A. Goldstein, Assessing the Accuracy of Ancestral Protein Reconstruction Methods, PLOS Computational Biology2, e69 (2006)
2006
-
[23]
Chantreau, C
M. Chantreau, C. Poux, M. F. Lensink, G. Brysbaert, X. Vekemans, and V. Castric, Asymmetrical diversifi- cation of the receptor-ligand interaction controlling self- incompatibility in Arabidopsis, eLife8, e50253 (2019)
2019
-
[24]
Dal´ en, P
L. Dal´ en, P. D. Heintzman, J. D. Kapp, and B. Shapiro, Deep-time paleogenomics and the limits of DNA survival, Science (New York, N.Y.)382, 48 (2023)
2023
-
[25]
M. C. Weiss, M. Preiner, J. C. Xavier, V. Zimorski, and W. F. Martin, The last universal common ancestor be- tween ancient Earth chemistry and the onset of genetics, PLoS Genetics14, e1007518 (2018)
2018
-
[26]
Fletcher and Z
W. Fletcher and Z. Yang, Indelible: a flexible simulator of biological sequence evolution, Molecular biology and evolution26, 1879 (2009)
2009
-
[27]
Ly-Trong, S
N. Ly-Trong, S. Naser-Khdour, R. Lanfear, and B. Q. Minh, Alisim: a fast and versatile phylogenetic sequence simulator for the genomic era, Molecular biology and evo- lution39, msac092 (2022)
2022
-
[28]
M. O. Dayhoff, R. M. Schwartz, and B. C. Orcutt, A model of evolutionary change in proteins, inAtlas of Pro- tein Sequence and Structure, Vol. 5, edited by M. O. Day- hoff (National Biomedical Research Foundation, Wash- ington, DC, 1978) pp. 345–352
1978
-
[29]
Whelan and N
S. Whelan and N. Goldman, A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach, Molecular Biol- ogy and Evolution18, 691 (2001)
2001
-
[30]
D. T. Jones, W. R. Taylor, and J. M. Thornton, The rapid generation of mutation data matrices from protein sequences, Bioinformatics8, 275 (1992)
1992
-
[31]
Trost, J
J. Trost, J. Haag, D. H¨ ohler, L. Jacob, A. Stamatakis, and B. Boussau, Simulations of Sequence Evolution: How (Un)realistic They Are and Why, Molecular Biology and Evolution41, msad277 (2024)
2024
-
[32]
J. A. G. De Visser and J. Krug, Empirical fitness land- scapes and the predictability of evolution, Nature Re- views Genetics15, 480 (2014)
2014
-
[33]
T. N. Starr and J. W. Thornton, Epistasis in protein evolution, Protein science25, 1204 (2016)
2016
-
[34]
M. S. Johnson, G. Reddy, and M. M. Desai, Epistasis and evolution: recent advances and an outlook for prediction, BMC biology21, 120 (2023)
2023
-
[35]
Domingo, P
J. Domingo, P. Baeza-Centurion, and B. Lehner, The causes and consequences of genetic interactions (epista- sis), Annual review of genomics and human genetics20, 433 (2019)
2019
-
[36]
J. Z. Chen, M. Bisardi, D. Lee, S. Cotogno, F. Zamponi, M. Weigt, and N. Tokuriki, Understanding epistatic net- works in the b1β-lactamases through coevolutionary sta- tistical modeling and deep mutational scanning, Nature communications15, 8441 (2024)
2024
-
[37]
X. Ding, Z. Zou, and C. L. Brooks, Deciphering pro- tein evolution and fitness landscapes with latent space models, Nature Communications10, 10.1038/s41467- 019-13633-0 (2019)
-
[38]
B. L. Hie, K. K. Yang, and P. S. Kim, Evolutionary ve- locity with protein language models predicts evolution- ary dynamics of diverse proteins, Cell Systems13, 274 (2022)
2022
-
[39]
Gorstein, M
E. Gorstein, M. Tang, H. Bruzzone, and C. Sol´ ıs-Lemus, Ancestral sequences cannot be accurately reconstructed via interpolation in a variational autoencoder’s latent 19 space (2025)
2025
-
[40]
Vaswani, N
A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. u. Kaiser, and I. Polosukhin, Attention is all you need, inAdvances in Neural Infor- mation Processing Systems, Vol. 30, edited by I. Guyon, U. V. Luxburg, S. Bengio, H. Wallach, R. Fergus, S. Vish- wanathan, and R. Garnett (Curran Associates, Inc., 2017)
2017
-
[41]
Koehl, S
A. Koehl, S. Prillo, M. Liu, J. Xiong, L. Weng, D. F. Savage, and Y. S. Song, Deep models of protein evolution in time generate realistic evolutionary trajectories and functional proteins (2026)
2026
-
[42]
De Leonardis, A
M. De Leonardis, A. Pagnani, and P. Barrat-Charlaix, Reconstruction of ancestral protein sequences using au- toregressive generative models, Molecular Biology and Evolution42, msaf070 (2025)
2025
-
[43]
L. Di Bari, T. Mora, A. Pagnani, A. M. Walczak, F. Zam- poni, and S. Rossi, Modeling protein evolution via gen- erative inference from monte carlo chains to population genetics, bioRxiv 10.64898/2026.02.09.704757 (2026)
-
[44]
L. D. Bari, M. Bisardi, S. Cotogno, M. Weigt, and F. Zamponi, Emergent time scales of epista- sis in protein evolution, Proceedings of the Na- tional Academy of Sciences121, e2406807121 (2024), https://www.pnas.org/doi/pdf/10.1073/pnas.2406807121
-
[45]
Bisardi, J
M. Bisardi, J. Rodriguez-Rivas, F. Zamponi, and M. Weigt, Modeling sequence-space exploration and emergence of epistatic signals in protein evolution, Molec- ular biology and evolution39, msab321 (2022)
2022
-
[46]
Rossi, L
S. Rossi, L. Di Bari, M. Weigt, and F. Zamponi, Fluctua- tions and the limit of predictability in protein evolution, Reports on Progress in Physics88, 078102 (2025)
2025
-
[47]
J. A. de la Paz, C. M. Nartey, M. Yuvaraj, and F. Mor- cos, Epistatic contributions promote the unification of incompatible models of neutral molecular evolution, Pro- ceedings of the National Academy of Sciences117, 5873 (2020)
2020
-
[48]
Alvarez, C
S. Alvarez, C. Nartey, N. Mercado, and F. Morcos, Novel sequence space explored by functional proteins generated through computational evolution-based design, Biophys- ical Journal121, 45a (2022)
2022
-
[49]
Weigt, R
M. Weigt, R. A. White, H. Szurmant, J. A. Hoch, and T. Hwa, Identification of direct residue contacts in protein–protein interaction by message passing, Proceed- ings of the National Academy of Sciences106, 67 (2009)
2009
-
[50]
W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat-Charlaix, M. Socolich, P. Kast, D. Hilvert, R. Monasson, S. Cocco, M. Weigt,et al., An evolution-based model for designing chorismate mutase enzymes, Science369, 440 (2020)
2020
-
[51]
Alvarez, C
S. Alvarez, C. M. Nartey, N. Mercado, J. A. de la Paz, T. Huseinbegovic, and F. Morcos, In vivo functional phe- notypes from a computational epistatic model of evolu- tion, Proceedings of the National Academy of Sciences 121, e2308895121 (2024)
2024
-
[52]
C. N. Lambert, V. Opuu, F. Calvanese, P. Pavlinova, F. Zamponi, E. J. Hayden, M. Weigt, M. Smerlak, and P. Nghe, Exploring the space of self-reproducing ri- bozymes using generative models, Nature communica- tions16, 7836 (2025)
2025
-
[53]
Expanding functional protein sequence space using high entropy generative models
R. Netti, E. Hinds, F. Calvanese, R. Ranganathan, M. Weigt, and F. Zamponi, Expanding functional pro- tein sequence space using high entropy generative mod- els, arXiv preprint arXiv:2605.03578 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[54]
Iqtree/iqtree3, iqtree (2025)
2025
-
[55]
Calvanese, M
F. Calvanese, M. Weigt, and P. Nghe, Generating ar- tificial ribozymes using sparse coevolutionary models, inRNA Design: Methods and Protocols, edited by A. Churkin and D. Barash (Springer US, New York, NY,
-
[56]
S. Q. Le, C. C. Dang, and O. Gascuel, Modeling protein evolution with several amino acid replacement matrices depending on site rates, Molecular Biology and Evolution 29, 2921 (2012)
2012
-
[57]
T. H. Jukes and C. R. Cantor, Evolution of Protein Molecules, inMammalian Protein Metabolism(Elsevier,
-
[58]
Price, Morgannprice/fasttree (2025)
M. Price, Morgannprice/fasttree (2025)
2025
-
[59]
Vigu´ e, G
L. Vigu´ e, G. Croce, M. Petitjean, E. Rupp´ e, O. Tenaillon, and M. Weigt, Deciphering polymorphism in 61,157 es- cherichia coli genomes via epistatic sequence landscapes, Nature Communications13, 4030 (2022)
2022
-
[60]
Pagnani and P
A. Pagnani and P. Barrat-Charlaix, Generative contin- uous time model reveals epistatic signatures in protein evolution, bioRxiv , 2025 (2025)
2025
-
[61]
E. R. Horta and M. Weigt, On the effect of phyloge- netic correlations in coevolution-based contact prediction in proteins, PLOS Computational Biology17, e1008957 (2021)
2021
-
[62]
Evans, C
W. Evans, C. Kenyon, Y. Peres, and L. J. Schulman, Broadcasting on Trees and the Ising Model, The Annals of Applied Probability10, 410 (2000), 2667156
2000
-
[63]
Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli, A. dos Santos Costa, M. Fazel-Zarandi, T. Sercu, S. Candido, and A. Rives, Evolutionary-scale prediction of atomic- level protein structure with a language model, Science 379, 1123 (2023)
2023
-
[64]
Jumper, R
J. Jumper, R. Evans, A. Pritzel, T. Green, M. Fig- urnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. ˇZ´ ıdek, A. Potapenko, A. Bridgland, C. Meyer, S. Kohl, A. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. S...
2021
-
[65]
Morcos, N
F. Morcos, N. P. Schafer, R. R. Cheng, J. N. Onuchic, and P. G. Wolynes, Coevolutionary information, protein folding landscapes, and the thermodynamics of natural selection, Proceedings of the National Academy of Sci- ences111, 12408 (2014)
2014
-
[66]
Veg/phylotree.js, iGEM/UCSD evolutionary biology and bioinformatics group (2025)
2025
-
[67]
Figliuzzi, P
M. Figliuzzi, P. Barrat-Charlaix, and M. Weigt, How Pairwise Coevolutionary Models Capture the Collective Residue Variability in Proteins?, Molecular Biology and Evolution35, 1018 (2018)
2018
- [68]
-
[69]
D. S. Marks, L. J. Colwell, R. Sheridan, T. A. Hopf, A. Pagnani, R. Zecchina, and C. Sander, Protein 3d structure computed from evolutionary sequence varia- tion, PLOS ONE6, 1 (2011)
2011
-
[70]
Rodriguez-Rivas, G
J. Rodriguez-Rivas, G. Croce, M. Muscat, and M. Weigt, Epistatic models predict mutable sites in sars-cov-2 pro- 20 teins and epitopes, Proceedings of the National Academy of Sciences119, e2113118119 (2022)
2022
-
[71]
Rodriguez Horta and M
E. Rodriguez Horta and M. Weigt, On the effect of phy- logenetic correlations in coevolution-based contact pre- diction in proteins, PLOS Computational Biology17, 1 (2021)
2021
-
[72]
Dietler, U
N. Dietler, U. Lupo, and A.-F. Bitbol, Impact of phy- logeny on structural contact inference from protein se- quence data, Journal of The Royal Society Interface20, 20220707 (2023)
2023
-
[73]
Vigu´ e and O
L. Vigu´ e and O. Tenaillon, Predicting the effect of mu- tations to investigate recent events of selection across 60,472 escherichia coli strains, Proceedings of the Na- tional Academy of Sciences120, e2304177120 (2023)
2023
-
[74]
Vigue, G
L. Vigue, G. Croce, M. Petitjean, E. Rupp´ e, O. Tenail- lon, and M. Weigt,Deciphering Polymorphism in 61,157 Escherichia Coli Genomes via Epistatic Sequence Land- scapes(2022)
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.