Recognition: unknown
Applications of deep generative models to DNA reaction kinetics and to cryogenic electron microscopy
Pith reviewed 2026-05-10 06:30 UTC · model grok-4.3
The pith
Biophysics-informed VAEs embed DNA reaction trajectories into interpretable pathways while GANs generate realistic cryo-EM density maps from protein structures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
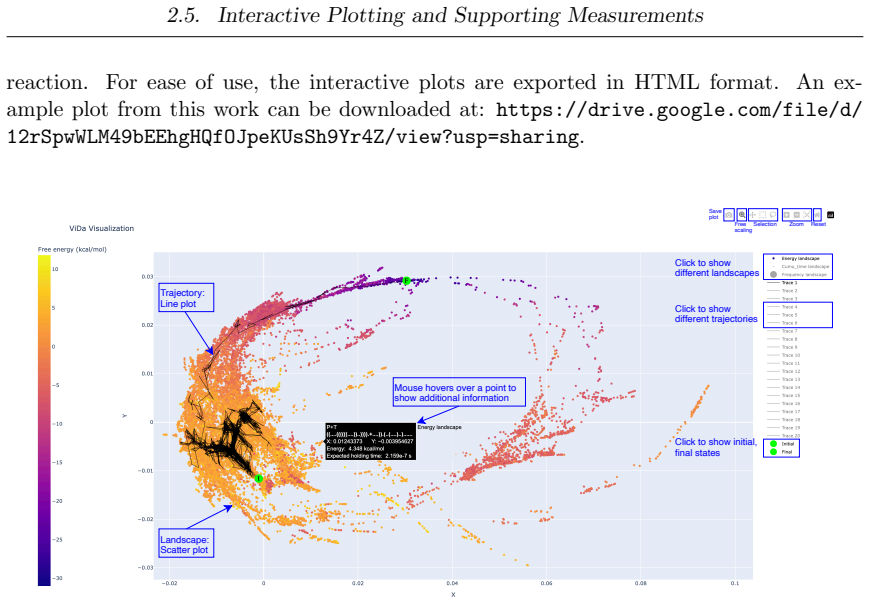
ViDa preserves structure in DNA reaction simulations by using biophysics-informed variational autoencoders and geometric scattering transforms to produce two-dimensional embeddings that cluster trajectory ensembles into reaction pathways, while Struc2mapGAN synthesizes high-fidelity experimental-like cryo-EM density maps from protein structures and CryoSAMU enhances intermediate-resolution maps through cross-attention integration of density features with structural embeddings.
What carries the argument
ViDa, the variational autoencoder with geometric scattering transforms for embedding DNA kinetics trajectories; Struc2mapGAN, the generative adversarial network for map synthesis; and CryoSAMU, the multimodal U-Net with cross-attention for map enhancement using protein embeddings.
If this is right
- DNA reaction simulation results become more interpretable through two-dimensional visualization and pathway clustering.
- New mechanistic insights into toehold-mediated strand displacement and hybridization reactions are obtained from the grouped trajectories.
- High-fidelity cryo-EM density maps can be generated directly from known protein structures for comparison with experiments.
- Intermediate-resolution cryo-EM maps are enhanced by integrating structural embeddings, aiding protein model building.
- Improved metrics and guidance are provided for evaluating deep learning methods in atomic model construction from cryo-EM data.
Where Pith is reading between the lines
- The embedding approach for simulations could extend to other molecular systems with known biophysical rules to improve interpretability.
- Generated and enhanced maps may reduce reliance on extensive new experiments by providing synthetic references for structure validation.
- Cross-attention fusion of pretrained protein models with experimental data suggests similar multimodal methods could apply to other imaging modalities in biology.
Load-bearing premise
The biophysics-informed embeddings accurately reflect true DNA reaction kinetics and the cross-attention fusion improves map quality without introducing artifacts.
What would settle it
Direct experimental validation showing that the reaction pathways clustered by ViDa fail to match observed DNA hybridization mechanisms or that Struc2mapGAN outputs differ systematically from real cryo-EM densities in blind tests.
Figures


























































read the original abstract
This dissertation explores how deep generative models can advance the analysis of challenging biological problems by integrating domain knowledge with deep learning. It focuses on two areas: DNA reaction kinetics and cryogenic electron microscopy (cryo-EM). In the first part, we present ViDa, a biophysics-informed framework leveraging variational autoencoders (VAEs) and geometric scattering transforms to generate biophysically-plausible embeddings of DNA reaction kinetics simulations. These embeddings are reduced to a two-dimensional space to visualize DNA hybridization and toehold-mediated strand displacement reactions. ViDa preserves structure and clusters trajectory ensembles into reaction pathways, making simulation results more interpretable and revealing new mechanistic insights. In the second part, we address key challenges in cryo-EM density map interpretation and protein structure modeling. We provide a comprehensive review and benchmarking of deep learning methods for atomic model building, with improved evaluation metrics and practical guidance. We then present Struc2mapGAN, a generative adversarial network that synthesizes high-fidelity experimental-like cryo-EM density maps from protein structures. Finally, we present CryoSAMU, a structure-aware multimodal U-Net that enhances intermediate-resolution cryo-EM maps by integrating density features with structural embeddings from protein language models via cross-attention. Overall, these contributions demonstrate the potential of deep generative models to interpret DNA reaction mechanisms and advance cryo-EM density map analysis and protein structure modeling.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. This dissertation applies deep generative models to DNA reaction kinetics and cryo-EM. It introduces ViDa, which combines variational autoencoders with geometric scattering transforms to produce 2D embeddings of DNA hybridization and toehold-mediated strand displacement simulation trajectories, claiming these embeddings preserve structure, cluster trajectories into reaction pathways, and yield new mechanistic insights. It also provides a review and benchmarking of deep learning methods for atomic model building in cryo-EM, introduces Struc2mapGAN to synthesize high-fidelity experimental-like density maps from protein structures, and presents CryoSAMU, a structure-aware multimodal U-Net that integrates density features with protein language model embeddings via cross-attention to enhance intermediate-resolution maps.
Significance. If the quantitative claims hold, ViDa could improve interpretability of molecular dynamics trajectories for nucleic acid reactions, while Struc2mapGAN and CryoSAMU could supply useful synthetic data and enhancement tools for cryo-EM structure determination. The biophysics-informed embedding approach and cross-attention integration represent a reasonable attempt to fuse domain knowledge with generative models. However, the absence of any reported metrics, baselines, or validation experiments in the abstract makes the practical significance difficult to assess at present.
major comments (3)
- [Abstract] Abstract: the central claims that ViDa 'preserves structure and clusters trajectory ensembles into reaction pathways' and 'revealing new mechanistic insights' are stated without any quantitative metrics, validation against known rate constants, transition-state ensembles, or baseline comparisons. This absence is load-bearing for the ViDa contribution.
- [ViDa framework] ViDa description: geometric scattering transforms are applied to time-series of DNA conformations, yet these operators are designed primarily for static point clouds and average over neighborhoods; the manuscript provides no explicit test showing that rare, high-energy transition configurations are retained rather than suppressed, which directly affects whether the observed 2D clusters reflect kinetic pathways or equilibrium structural similarity.
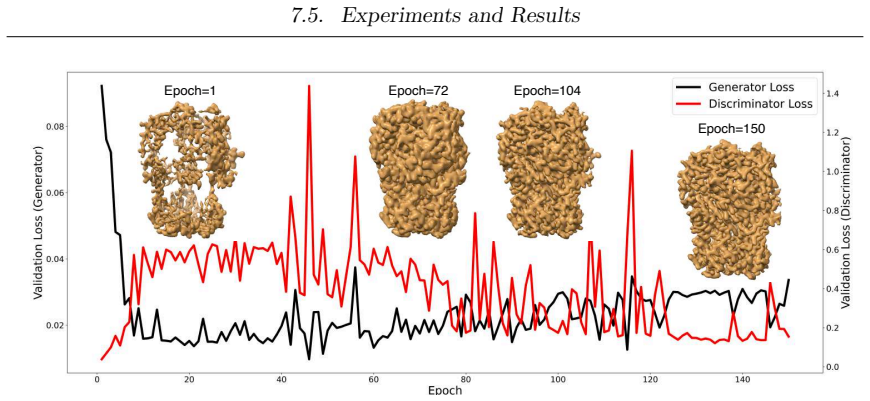
- [Cryo-EM contributions] Struc2mapGAN and CryoSAMU sections: assertions of 'high-fidelity experimental-like' maps and 'meaningfully improves map quality' lack reported quantitative measures such as FSC curves, resolution estimates, or artifact quantification against experimental references, which are required to support the generative modeling claims.
minor comments (1)
- [Benchmarking review] The benchmarking section for atomic model building would be strengthened by explicit listing of the improved evaluation metrics and the precise criteria used for 'practical guidance'.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our dissertation. We respond to each major comment below and indicate the revisions we will make to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claims that ViDa 'preserves structure and clusters trajectory ensembles into reaction pathways' and 'revealing new mechanistic insights' are stated without any quantitative metrics, validation against known rate constants, transition-state ensembles, or baseline comparisons. This absence is load-bearing for the ViDa contribution.
Authors: The abstract provides a high-level overview of the work, while the detailed quantitative metrics, including clustering performance, comparisons to known rate constants, and validation of transition states, are presented in the main body of the dissertation. To address the referee's concern, we will revise the abstract to include brief references to these quantitative validations. revision: yes
-
Referee: [ViDa framework] ViDa description: geometric scattering transforms are applied to time-series of DNA conformations, yet these operators are designed primarily for static point clouds and average over neighborhoods; the manuscript provides no explicit test showing that rare, high-energy transition configurations are retained rather than suppressed, which directly affects whether the observed 2D clusters reflect kinetic pathways or equilibrium structural similarity.
Authors: We note that the geometric scattering transforms are used to extract multi-scale features from each conformation in the trajectory, and the variational autoencoder is trained on the full time-series to capture temporal dynamics. Although an explicit test for retention of rare high-energy configurations is not detailed in the current manuscript, the 2D embeddings demonstrate clustering consistent with known kinetic pathways rather than just equilibrium structures. We will add an explicit analysis or discussion of this point in the revised manuscript. revision: partial
-
Referee: [Cryo-EM contributions] Struc2mapGAN and CryoSAMU sections: assertions of 'high-fidelity experimental-like' maps and 'meaningfully improves map quality' lack reported quantitative measures such as FSC curves, resolution estimates, or artifact quantification against experimental references, which are required to support the generative modeling claims.
Authors: Quantitative measures including FSC curves, resolution estimates, and comparisons to experimental references are provided in the results sections for both Struc2mapGAN and CryoSAMU. We will revise the manuscript to more prominently feature these metrics, perhaps with a dedicated table, to better support the claims. revision: yes
Circularity Check
No circularity: descriptive application of existing generative models with no load-bearing derivations or self-referential reductions.
full rationale
The manuscript presents ViDa (VAE + geometric scattering for DNA trajectory embeddings), Struc2mapGAN, and CryoSAMU as applied frameworks without exhibiting any mathematical derivation chains, equations, or fitted-parameter predictions that reduce to their own inputs by construction. No self-citations, uniqueness theorems, or ansatzes are invoked in a load-bearing way; the work is empirical and architectural rather than deductive. The central claims rest on model performance and visualization rather than any step that is definitionally equivalent to its premise.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
The abtem code: transmission electron microscopy from first principles.Open Research Europe, 1:24, 2021
Jacob Madsen and Toma Susi. The abtem code: transmission electron microscopy from first principles.Open Research Europe, 1:24, 2021
2021
-
[2]
Image formation modeling in cryo-electron microscopy.Journal of structural biology, 183(1):19–32, 2013
Miloˇ s Vulovi´ c, Raimond BG Ravelli, Lucas J van Vliet, Abraham J Koster, Ivan Lazi´ c, Uwe L¨ ucken, Hans Rullg˚ ard, Ozan¨Oktem, and Bernd Rieger. Image formation modeling in cryo-electron microscopy.Journal of structural biology, 183(1):19–32, 2013
2013
-
[3]
Could egg white lysozyme be solved by single particle cryo-em?Journal of chemical information and modeling, 60(5):2605–2613, 2020
Yue Zhang, R Tammaro, Peter J Peters, and RBG Ravelli. Could egg white lysozyme be solved by single particle cryo-em?Journal of chemical information and modeling, 60(5):2605–2613, 2020
2020
-
[4]
Film: Visual reasoning with a general conditioning layer
Ethan Perez, Florian Strub, Harm De Vries, Vincent Dumoulin, and Aaron Courville. Film: Visual reasoning with a general conditioning layer. InProceedings of the AAAI conference on artificial intelligence, volume 32, pages 3942–3951, 2018
2018
-
[5]
Single-particle cryo-em—how did it get here and where will it go
Yifan Cheng. Single-particle cryo-em—how did it get here and where will it go. Science, 361(6405):876–880, 2018
2018
-
[6]
Principles of cryo-em single-particle image processing.Journal of Electron Microscopy, 65(1):57–67, 2015
Fred J Sigworth. Principles of cryo-em single-particle image processing.Journal of Electron Microscopy, 65(1):57–67, 2015
2015
-
[7]
Jochen B¨ ohm, Achilleas S Frangakis, Reiner Hegerl, Stephan Nickell, Dieter Typke, and Wolfgang Baumeister. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms.Proceedings of the National Academy of Sciences, 97(26):14245–14250, 2000
2000
-
[8]
Automatic local resolution-based sharpening of cryo-em maps.Bioin- formatics, 36(3):765–772, 2020
Erney Ram´ ırez-Aportela, Jose Luis Vilas, Alisa Glukhova, Roberto Melero, Pablo Conesa, Marta Mart´ ınez, David Maluenda, Javier Mota, Amaya Jim´ enez, Javier Vargas, et al. Automatic local resolution-based sharpening of cryo-em maps.Bioin- formatics, 36(3):765–772, 2020
2020
-
[9]
Atypical structural snapshots of human cytomegalovirus gpcr interactions with host g proteins.Science advances, 8(3):eabl5442, 2022
Naotaka Tsutsumi, Shoji Maeda, Qianhui Qu, Martin V¨ ogele, Kevin M Jude, Carl- Mikael Suomivuori, Ouliana Panova, Deepa Waghray, Hideaki E Kato, Andrew Ve- lasco, et al. Atypical structural snapshots of human cytomegalovirus gpcr interactions with host g proteins.Science advances, 8(3):eabl5442, 2022
2022
-
[10]
Sigmoid-weighted linear units for neural network function approximation in reinforcement learning.Neural networks, 107:3– 11, 2018
Stefan Elfwing, Eiji Uchibe, and Kenji Doya. Sigmoid-weighted linear units for neural network function approximation in reinforcement learning.Neural networks, 107:3– 11, 2018
2018
-
[11]
Trans- formers are rnns: Fast autoregressive transformers with linear attention
Angelos Katharopoulos, Apoorv Vyas, Nikolaos Pappas, and Fran¸ cois Fleuret. Trans- formers are rnns: Fast autoregressive transformers with linear attention. InInterna- tional conference on machine learning, pages 5156–5165. PMLR, 2020. 132
2020
-
[12]
Auxiliary modality learning with generalized curriculum distillation
Yu Shen, Xijun Wang, Peng Gao, and Ming Lin. Auxiliary modality learning with generalized curriculum distillation. InInternational Conference on Machine Learn- ing, pages 31057–31076. PMLR, 2023
2023
-
[13]
Decoupled Weight Decay Regularization
Ilya Loshchilov and Frank Hutter. Decoupled weight decay regularization.arXiv preprint arXiv:1711.05101, 2017
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[14]
New tools for the analysis and validation of cryo-em maps and atomic models.Acta Crystallographica Section D: Structural Biology, 74(9):814–840, 2018
Pavel V Afonine, Bruno P Klaholz, Nigel W Moriarty, Billy K Poon, Oleg V Sobolev, Thomas C Terwilliger, Paul D Adams, and Alexandre Urzhumtsev. New tools for the analysis and validation of cryo-em maps and atomic models.Acta Crystallographica Section D: Structural Biology, 74(9):814–840, 2018
2018
-
[15]
Structure of the transporter associated with antigen processing trapped by herpes simplex virus.Elife, 5:e21829, 2016
Michael L Oldham, Nikolaus Grigorieff, and Jue Chen. Structure of the transporter associated with antigen processing trapped by herpes simplex virus.Elife, 5:e21829, 2016
2016
-
[16]
Tir-catalyzed adp-ribosylation re- actions produce signaling molecules for plant immunity.Science, 377(6605):eabq8180, 2022
Aolin Jia, Shijia Huang, Wen Song, Junli Wang, Yonggang Meng, Yue Sun, Lina Xu, Henriette Laessle, Jan Jirschitzka, Jiao Hou, et al. Tir-catalyzed adp-ribosylation re- actions produce signaling molecules for plant immunity.Science, 377(6605):eabq8180, 2022
2022
-
[17]
Structure of human dispatched-1 pro- vides insights into hedgehog ligand biogenesis.Life science alliance, 3(8), 2020
Hongwen Chen, Yang Liu, and Xiaochun Li. Structure of human dispatched-1 pro- vides insights into hedgehog ligand biogenesis.Life science alliance, 3(8), 2020
2020
-
[18]
Computational design of transmembrane pores.Nature, 585(7823):129–134, 2020
Chunfu Xu, Peilong Lu, Tamer M Gamal El-Din, Xue Y Pei, Matthew C Johnson, Atsuko Uyeda, Matthew J Bick, Qi Xu, Daohua Jiang, Hua Bai, et al. Computational design of transmembrane pores.Nature, 585(7823):129–134, 2020
2020
-
[19]
Jun-Jie Liu, Chu-Ya Niu, Yao Wu, Dan Tan, Yang Wang, Ming-Da Ye, Yang Liu, Wenwei Zhao, Ke Zhou, Quan-Sheng Liu, et al. Cryoem structure of yeast cytoplas- mic exosome complex.Cell research, 26(7):822–837, 2016. 133 Appendix A Supplementary Materials for Chapter 1 A.1 Dot-Parenthesis Notation Dot-parenthesis (dp) notation is a simple way to represent a ps...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.