Recognition: unknown
What Makes a Bacterial Model a Good Reservoir Computer? Predicting Performance from Separability and Similarity
Pith reviewed 2026-05-10 07:49 UTC · model grok-4.3
The pith
Bacterial metabolic models can perform nonlinear classification tasks when their growth curves serve as reservoir states.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Bacterial metabolic dynamics simulated by dynamic flux balance analysis can act as effective physical reservoirs. Growth curves generated from glucose and xylose inputs provide state trajectories that a linear readout can use to classify nonlinear patterns at high accuracy. Wild-type E. coli outperforms all its single-gene deletion mutants, while different species trade off speed of convergence against maximum accuracy. The difference between kernel rank and generalization rank of the growth-curve matrices is associated with better performance, although deviations and sensitivity at low ranks limit reliable prediction.
What carries the argument
The difference between kernel rank and generalization rank extracted from the matrix of simulated growth curves, which quantifies how the metabolic dynamics separate distinct inputs while preserving enough similarity for generalization.
If this is right
- Strains whose growth curves produce large differences between kernel and generalization ranks are expected to deliver higher classification accuracy.
- Single-gene deletions in E. coli systematically reduce the dynamical richness needed for reservoir performance relative to the wild type.
- Species-level differences create a practical trade-off: some converge quickly while others reach higher peak accuracy.
- Bacterial metabolic models constitute viable substrates for physical reservoir computing and can be screened in simulation before experimental use.
Where Pith is reading between the lines
- Metabolic models could be further screened or engineered by varying additional nutrients or regulatory parameters to target specific computational properties.
- The same separability-similarity measures might be applied to other biological substrates such as yeast or mammalian cells to identify additional computing candidates.
- If the rank-difference predictor holds under richer inputs, it could guide the selection of microbes for real-world sensing or control tasks rather than only synthetic benchmarks.
Load-bearing premise
Simulated growth curves produced by dynamic flux balance analysis with only two sugar inputs contain the dynamical richness that living bacteria would exhibit, and success on synthetic random tasks will translate to useful performance on natural inputs.
What would settle it
An experiment in which real bacterial cultures exposed to varying glucose-xylose sequences produce growth curves whose state matrix yields linear-readout accuracy no better than chance on the same classification tasks would falsify the claim that these metabolic dynamics support nonlinear computation.
Figures

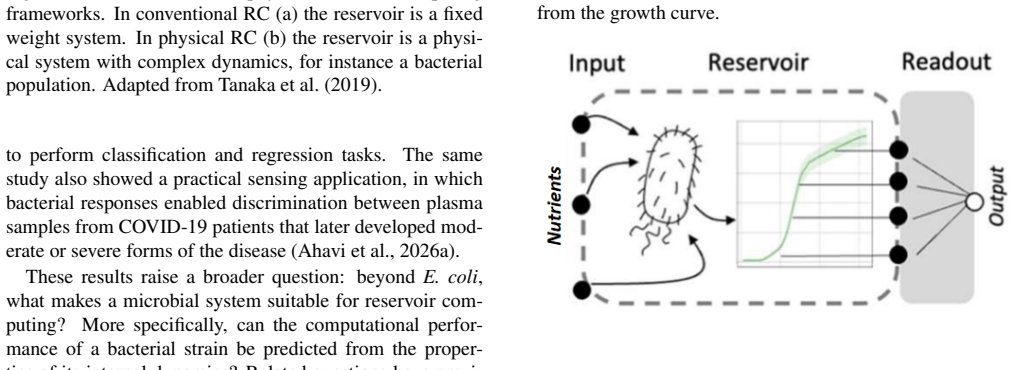








read the original abstract
Biological systems are promising substrates for computation because they naturally process environmental information through complex internal dynamics. In this study, we investigate whether bacterial metabolic models can act as physical reservoirs and whether their computational performance can be predicted from dynamical properties linked to separability and similarity. We simulated the growth dynamics of five bacterial species, one yeast species, and 29 Escherichia coli single-gene deletion mutants using dynamic flux balance analysis (dFBA), with glucose and xylose concentrations as inputs and growth curves as reservoir states. Computational performance was assessed on random nonlinear classification tasks using a linear readout, while reservoir properties linked to separability and similarity were characterised through kernel and generalisation ranks computed from growth-curve state matrices. Several microbial models achieved high classification accuracy, showing that bacterial metabolic dynamics can support nonlinear computation. Clear differences were observed between species, with some models converging more rapidly and others reaching higher maximum accuracy, revealing a trade-off between convergence speed and peak performance. In contrast, all E. coli mutants were dominated by the wild-type model, suggesting that gene deletions reduce the dynamical richness required for efficient computation. The difference between kernel and generalisation ranks was generally associated with improved accuracy, but deviations across models and sensitivity at low rank values limited its predictive power in practice. Overall, these results show that bacterial metabolic models constitute promising substrates for reservoir computing and provide a first step towards identifying microbial strains with favourable computational properties for future experimental implementations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses dynamic flux balance analysis (dFBA) to simulate growth dynamics of five bacterial species, one yeast, and 29 E. coli single-gene deletion mutants, with glucose and xylose concentrations as inputs. Growth curves serve as reservoir states for nonlinear classification tasks solved by a linear readout. Kernel and generalization ranks are computed from the resulting state matrices to characterize separability and similarity, and their difference is examined as a predictor of classification accuracy. Several models achieve high accuracy, wild-type E. coli outperforms its mutants, species show trade-offs in convergence speed versus peak performance, and rank differences are generally but noisily associated with better accuracy, with limited predictive power overall.
Significance. If the simulation results hold, the work demonstrates that bacterial metabolic dynamics can support nonlinear reservoir computing and identifies preliminary links between dynamical properties and performance. This provides a first step toward selecting microbial strains for experimental bio-computing applications, with the breadth of models (including mutants) adding value. The explicit scoping to simulations and acknowledgment of the rank metric's limited power are appropriate strengths.
major comments (2)
- [Methods and Results (rank-accuracy association)] The kernel and generalization ranks are computed directly from the same growth-curve state matrices used to train and test the linear readout (Methods and Results sections on rank calculation and performance evaluation). This data dependence makes the reported association with accuracy post-hoc and circular rather than an independent prediction, which is load-bearing for the title's claim of 'Predicting Performance from Separability and Similarity' and the abstract's framing of the rank difference as a performance predictor. The manuscript should either recompute ranks on held-out data, provide a non-circular formulation, or reframe the association as correlative only.
- [Results] No error bars, standard deviations from multiple runs, or statistical tests (e.g., correlation coefficients with p-values) are provided for the classification accuracies or the rank-difference associations across models and tasks (Results section on performance and rank differences). This weakens evaluation of the reported species differences, mutant underperformance relative to wild-type, and the 'generally associated but noisy' rank link, especially given the limited predictive power noted by the authors.
minor comments (2)
- [Methods] The exact mathematical definitions of kernel rank and generalization rank (e.g., how the state matrices are processed to obtain each) should be stated with equations in the Methods section for clarity and reproducibility.
- [Discussion] The manuscript would benefit from a brief discussion of how the choice of only glucose/xylose inputs and synthetic random classification tasks might limit extrapolation to natural environmental inputs or real biological computation.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed review. We address each major comment below, indicating the revisions we will incorporate to improve the manuscript.
read point-by-point responses
-
Referee: [Methods and Results (rank-accuracy association)] The kernel and generalization ranks are computed directly from the same growth-curve state matrices used to train and test the linear readout (Methods and Results sections on rank calculation and performance evaluation). This data dependence makes the reported association with accuracy post-hoc and circular rather than an independent prediction, which is load-bearing for the title's claim of 'Predicting Performance from Separability and Similarity' and the abstract's framing of the rank difference as a performance predictor. The manuscript should either recompute ranks on held-out data, provide a non-circular formulation, or reframe the association as correlative only.
Authors: We acknowledge that the kernel and generalization ranks are computed from the same state matrices as those used for training and evaluating the linear readout, which renders the observed link an in-sample association rather than an independent prediction on held-out data. The current abstract already qualifies the rank difference as having 'limited predictive power,' consistent with this limitation. To address the concern directly, we will add explicit clarification in the Methods and Results sections stating that the association is correlative and derived from the same simulations. We will also revise the abstract to emphasize 'associations with performance' in place of stronger predictive language. The title frames the central research question rather than asserting a validated predictive method; we will retain it but ensure the body text aligns with the correlative nature of the analysis performed. We note that recomputing ranks on fully held-out data would require a substantial redesign of the experimental protocol and is left for future work. revision: partial
-
Referee: [Results] No error bars, standard deviations from multiple runs, or statistical tests (e.g., correlation coefficients with p-values) are provided for the classification accuracies or the rank-difference associations across models and tasks (Results section on performance and rank differences). This weakens evaluation of the reported species differences, mutant underperformance relative to wild-type, and the 'generally associated but noisy' rank link, especially given the limited predictive power noted by the authors.
Authors: We agree that the absence of variability measures and statistical tests weakens the presentation of species-level differences and the rank-accuracy relationship. In the revised manuscript we will rerun the classification tasks across multiple independent realizations using different random seeds for task generation and input sequences. From these runs we will report standard deviations and error bars on the accuracy values and compute Pearson or Spearman correlation coefficients (with p-values) between rank differences and accuracies. These additions will allow readers to assess the reliability of the reported trends, including the dominance of wild-type E. coli over its mutants and the noisy but generally positive rank link. revision: yes
Circularity Check
No significant circularity
full rationale
The paper performs an empirical investigation by running dFBA simulations to generate growth-curve state matrices, then measuring both classification accuracy (via linear readout on synthetic tasks) and kernel/generalisation ranks from those same matrices, followed by reporting an observed association with explicit caveats on its limited predictive power. No derivation chain is claimed that reduces a result to its inputs by construction, no parameters are fitted and then relabeled as predictions, and no self-citation or uniqueness theorem is invoked to force the central claims. The work is scoped to the simulated models and presents the rank-accuracy link as an empirical observation rather than an independent first-principles prediction, making the analysis self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Linear readout is sufficient to extract computational performance from reservoir states
- domain assumption dFBA growth dynamics with glucose/xylose inputs capture relevant separability and similarity properties
Reference graph
Works this paper leans on
-
[1]
A., Palsson, B
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). Cobrapy: Constraints-based reconstruction and anal- ysis for python.BMC Systems Biology, 7(1)
2013
-
[2]
Elowitz, M. B. and Leibler, S. (2000). A synthetic os- cillatory network of transcriptional regulators.Nature, 403(6767):335–338
2000
-
[3]
S., Cantor, C
Gardner, T. S., Cantor, C. R., and Collins, J. J. (2000). Construc- tion of a genetic toggle switch in escherichia coli.Nature, 403(6767):339–342
2000
-
[4]
Henson, M. A. and Hanly, T. J. (2014). Dynamic flux balance analysis for synthetic microbial communities.IET Systems Biology, 8(5):214–229
2014
-
[5]
IEC, O., IFCC, R., ILAC, L., ISO, L., IUPAC, M., IUPAP, R., et al. (1995). Guide to the expression of uncertainty in measure- ment.ISO, Geneva
1995
-
[6]
A., Islam, K
Kaplan, N. A., Islam, K. N., Kanis, F. C., Verderber, J. R., Wang, X., Jones, J. A., and Koffas, M. A. (2024). Simultaneous glu- cose and xylose utilization by anEscherichia colicatabolite repression mutant.Applied and Environmental Microbiology, 90(2)
2024
-
[7]
Lauer, F. (2014). Vc-dimension of hyperplanes.https:// mlweb.loria.fr/book/en/VCdimhyperplane. html(Accessed: 2026-04-10)
2014
-
[8]
and Maass, W
Legenstein, R. and Maass, W. (2007). Edge of chaos and predic- tion of computational performance for neural circuit models. Neural Networks, 20(3):323–334. Lukoˇseviˇcius, M. and Jaeger, H. (2009). Reservoir computing ap- proaches to recurrent neural network training.Computer sci- ence review, 3(3):127–149
2007
-
[9]
S., and Doyle, F
Mahadevan, R., Edwards, J. S., and Doyle, F. J. (2002). Dynamic flux balance analysis of diauxic growth in escherichia coli. Biophysical Journal, 83(3):1331–1340
2002
-
[10]
Nakajima, K. (2020). Physical reservoir computing—an intro- ductory perspective.Japanese Journal of Applied Physics, 59(6):060501
2020
-
[11]
Brucher, M., Perrot, M., and Duchesnay, E. (2011). Scikit- learn: Machine learning in Python.Journal of Machine Learning Research, 12:2825–2830
2011
-
[12]
Seoane, L. F. (2019). Evolutionary aspects of reservoir computing. Philosophical Transactions of the Royal Society B: Biological Sciences, 374(1774):20180377
2019
-
[13]
Takeda, S., Numata, H., Nakano, D., and Hirose, A. (2019). Recent advances in physical reservoir computing: A review. Neural Networks, 115:100–123
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.