Recognition: 3 theorem links
· Lean TheoremProtDBench: A Unified Benchmark of Protein Binder Design and Evaluation
Pith reviewed 2026-05-08 18:09 UTC · model grok-4.3
The pith
A new benchmark for protein binder design shows that common structure prediction models disagree substantially on which designs succeed under identical rules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
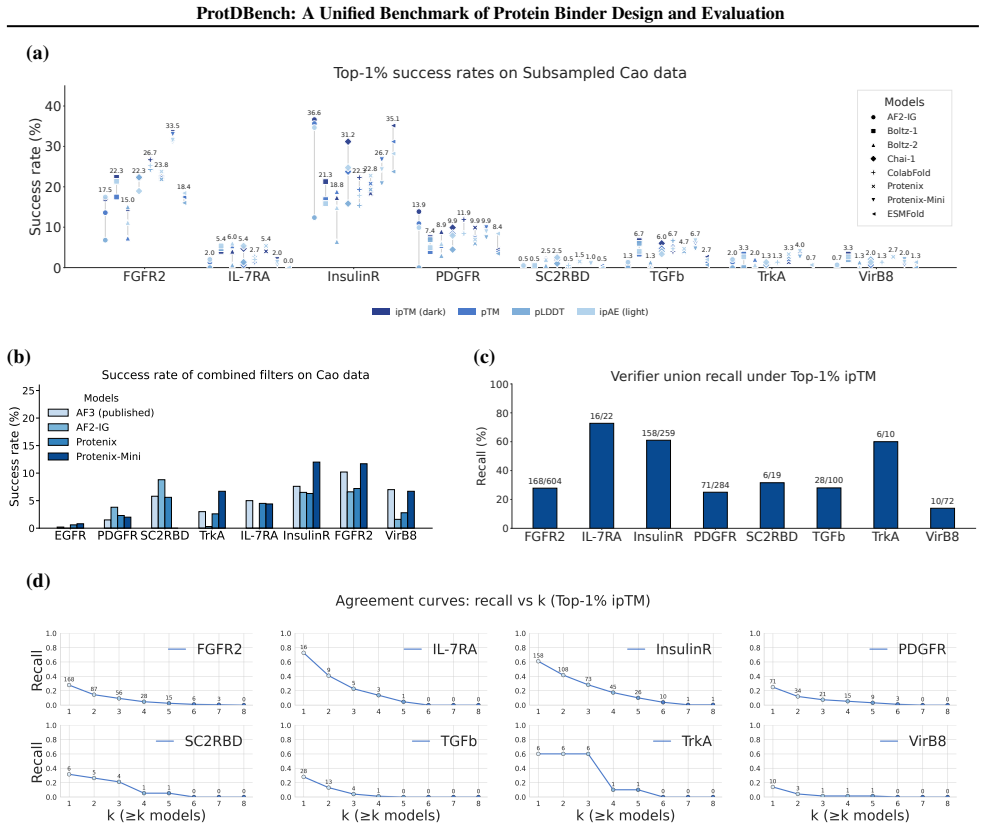
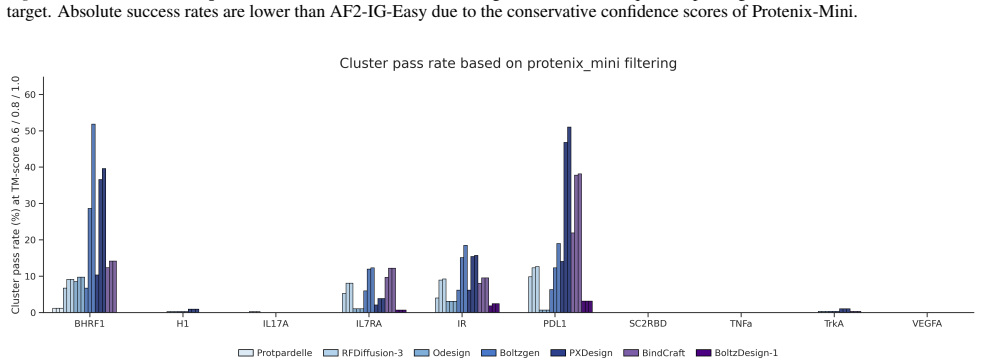
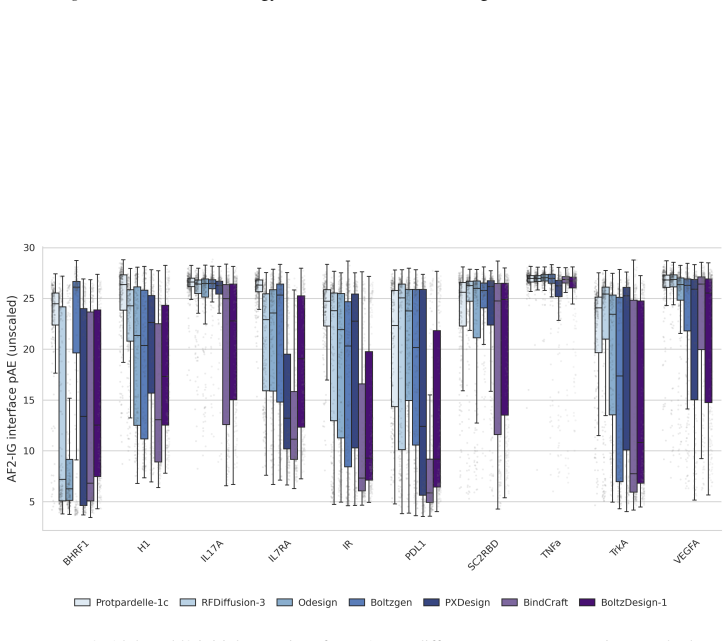
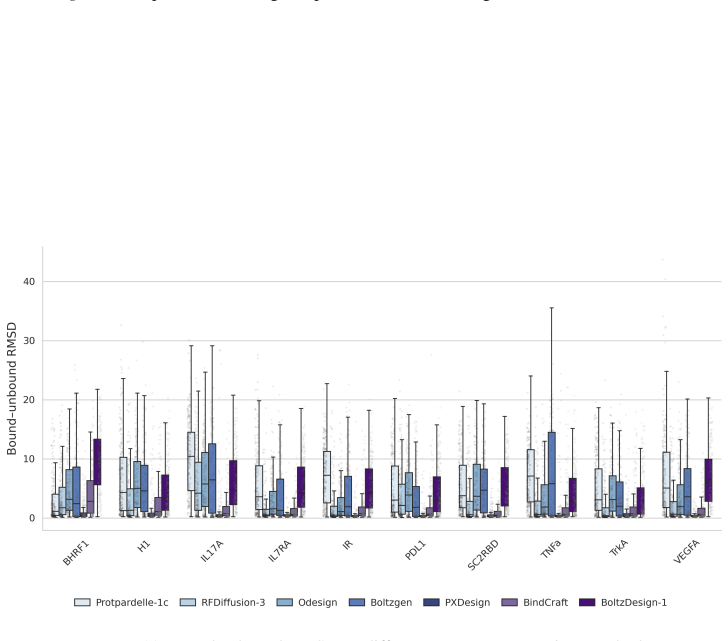
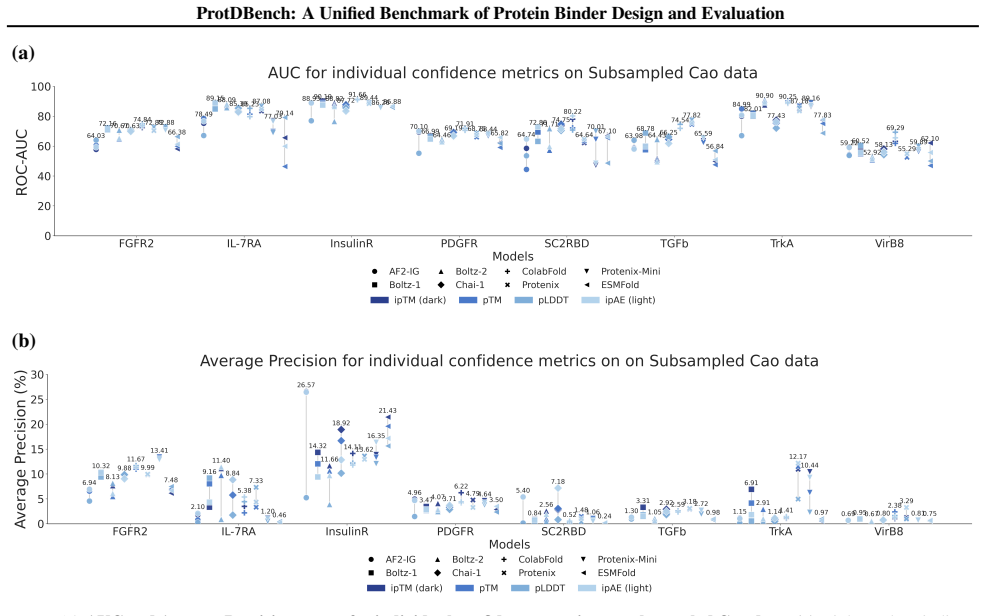
ProtDBench defines unified benchmark tasks, evaluation protocols, and success criteria for protein binder design. Using a large wet-lab annotated dataset, analysis of structure prediction models as verifiers reveals substantial verifier-dependent bias and limited agreement under identical filtering protocols. Benchmarking of representative generative methods across ten targets under a fixed protocol exposes systematic differences induced by filtering rules, success definitions, and throughput-aware evaluation between computational efficiency, success rate, and structural diversity.
What carries the argument
The ProtDBench evaluation framework, which specifies unified tasks, protocols, success criteria, 24-hour throughput metrics, and cluster-level criteria for structural diversity.
If this is right
- The same set of designed sequences receives different success labels depending on which structure prediction model acts as verifier.
- The measured performance of any given design method shifts when filtering protocols or success definitions change.
- Throughput-aware metrics under a fixed time budget expose trade-offs between the number of designs produced and their success rates.
- Cluster-level success criteria add a requirement for structural diversity beyond per-sequence success.
Where Pith is reading between the lines
- Design methods might be optimized to satisfy the preferences of the specific verifiers in the benchmark rather than actual binding behavior.
- Expanding the set of targets or incorporating newer structure predictors could change the observed levels of verifier disagreement.
- Reporting results under multiple verifier settings would help show whether a design method is robust to evaluation choices.
Load-bearing premise
That the wet-lab annotated dataset together with the chosen success criteria and filtering protocols accurately reflect real experimental binder performance.
What would settle it
Applying the same analysis to an independent collection of wet-lab validated binders and finding that the structure prediction models agree more closely or produce different method rankings than those reported.
Figures







read the original abstract
Recent advances in de novo protein binder design have enabled increasing experimental validation, yet reported in silico metrics remain difficult to interpret or compare across studies due to non-standardized evaluation protocols. We introduce ProtDBench, a standardized and throughput-aware evaluation framework for protein binder design. ProtDBench defines unified benchmark tasks, evaluation protocols, and success criteria, enabling systematic analysis of how evaluation design influences observed performance. Using a large wet-lab annotated dataset, we analyze commonly used structure prediction models as evaluation verifiers, revealing substantial verifier-dependent bias and limited agreement under identical filtering protocols. We then benchmark representative open-source generative binder design methods across ten diverse protein targets under a fixed evaluation protocol. Beyond per-sequence success rates, ProtDBench incorporates throughput-aware metrics based on a fixed 24-hour budget, as well as cluster-level success criteria to account for structural diversity. Together, these results expose systematic differences induced by filtering rules, success definitions, and throughput-aware evaluation between computational efficiency, success rate, and structural diversity. Overall, ProtDBench provides a fair and reproducible evaluation pipeline that supports systematic and controlled comparison of protein binder design methods under realistic evaluation settings.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces ProtDBench, a standardized benchmark framework for de novo protein binder design that defines unified tasks, evaluation protocols, success criteria, and throughput-aware metrics. Using a large wet-lab annotated dataset, it demonstrates substantial verifier-dependent bias and limited agreement among structure prediction models under identical filters, then benchmarks representative generative methods across ten targets while incorporating cluster-level diversity criteria.
Significance. If the wet-lab dataset and protocols hold as reliable proxies, ProtDBench would fill a critical gap by enabling reproducible, controlled comparisons of binder design methods and exposing how filtering rules and verifier choice systematically alter reported performance, success rates, and efficiency rankings. The throughput budget and diversity metrics are particularly valuable additions for practical assessment.
major comments (2)
- [Abstract and dataset description] The central claim that ProtDBench supplies a fair pipeline under realistic settings rests on the wet-lab annotated dataset serving as ground truth; however, the manuscript provides no external validation, correlation analysis with independent affinity measurements, or sensitivity checks for target selection and annotation quality biases (see abstract and the dataset description section).
- [Evaluation protocols and results sections] § on success criteria and filtering protocols: the definitions of success (structure-based or otherwise) and the 24-hour throughput budget are presented without reported correlation to actual experimental binding outcomes, which directly affects the validity of the observed verifier biases and method rankings.
minor comments (2)
- [Introduction] The abstract and introduction would benefit from explicit citation of prior non-standardized evaluation practices in the field to better motivate the need for unification.
- [Figures and tables] Figure legends and table captions should clarify the exact number of sequences per target and the precise definition of 'cluster-level success' to improve reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback. We address each major comment below and outline revisions to improve clarity on dataset reliability and the grounding of success criteria.
read point-by-point responses
-
Referee: [Abstract and dataset description] The central claim that ProtDBench supplies a fair pipeline under realistic settings rests on the wet-lab annotated dataset serving as ground truth; however, the manuscript provides no external validation, correlation analysis with independent affinity measurements, or sensitivity checks for target selection and annotation quality biases (see abstract and the dataset description section).
Authors: We agree that the manuscript's central claims depend on the wet-lab dataset serving as a reliable proxy for ground truth. The dataset aggregates binders from published experimental studies with wet-lab validation (e.g., via SPR, ELISA, or functional assays). However, the current version does not include new external validation, cross-correlations with independent affinity data, or formal sensitivity checks. We will revise the dataset description section to expand on data provenance, annotation quality controls, known limitations, and a sensitivity analysis for target selection and annotation biases. This will better substantiate the fairness of the evaluation pipeline. revision: partial
-
Referee: [Evaluation protocols and results sections] § on success criteria and filtering protocols: the definitions of success (structure-based or otherwise) and the 24-hour throughput budget are presented without reported correlation to actual experimental binding outcomes, which directly affects the validity of the observed verifier biases and method rankings.
Authors: The success criteria and 24-hour throughput budget are defined using computational proxies (structure prediction agreement, sequence and cluster diversity) with the wet-lab annotations serving as the reference for positive binders. We acknowledge that the manuscript does not report explicit quantitative correlations between these in silico definitions and independent experimental binding outcomes, which could influence interpretation of the verifier biases and method rankings. We will revise the evaluation protocols section to explicitly discuss the proxy nature of the metrics, reference any available supporting literature on their correlation with experiments, and add a limitations paragraph. This will strengthen the presentation without altering the core results. revision: partial
Circularity Check
No circularity: benchmark definitions and analyses are independent of self-referential inputs
full rationale
The paper introduces ProtDBench by defining new unified benchmark tasks, evaluation protocols, success criteria, and throughput-aware metrics applied to a wet-lab annotated dataset. It then uses these definitions to analyze verifier biases in structure prediction models and to benchmark generative design methods across targets. No load-bearing claims reduce by construction to fitted parameters, self-citations, or renamed inputs; the central results on bias, agreement, and method comparisons follow directly from applying the externally grounded dataset and fixed protocols. This is a methods/benchmark paper with no derivation chain that collapses to its own definitions.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Structure prediction models serve as appropriate proxies for experimental validation of binder designs.
- domain assumption The wet-lab annotated dataset provides reliable ground truth for benchmarking.
Lean theorems connected to this paper
-
Cost.FunctionalEquation / Foundation.AlphaCoordinateFixationwashburn_uniqueness_aczel; alpha_pin_under_high_calibration unclearFilter thresholds: binder ipTM>0.85, binder pTM>0.88, complex RMSD<2.5Å (grid-searched on Cao dataset)
Reference graph
Works this paper leans on
-
[1]
Geoflow-v2: A unified atomic diffusion model for protein structure prediction and de novo de- sign.bioRxiv, pp
BioGeometry. Geoflow-v2: A unified atomic diffusion model for protein structure prediction and de novo de- sign.bioRxiv, pp. 2025–05,
2025
-
[2]
Butcher, J., Krishna, R., Mitra, R., Brent, R. I., Li, Y ., Corley, N., Kim, P., Funk, J., Mathis, S., Sa- like, S., Muraishi, A., Eisenach, H., Thompson, T. R., Chen, J., Politanska, Y ., Sehgal, E., Coven- try, B., Zhang, O., Qiang, B., Didi, K., Kazman, M., DiMaio, F., and Baker, D. De novo design of all-atom biomolecular interactions with rfdiffusion3...
-
[3]
Chai- 1: Decoding the molecular interactions of life.BioRxiv, pp
ChaiDiscovery, Boitreaud, J., Dent, J., McPartlon, M., Meier, J., Reis, V ., Rogozhonikov, A., and Wu, K. Chai- 1: Decoding the molecular interactions of life.BioRxiv, pp. 2024–10,
2024
-
[4]
Zero-shot antibody design in a 24-well plate.bioRxiv, pp
ChaiDiscovery, Boitreaud, J., Dent, J., Geisz, D., McPart- lon, M., Meier, J., Qiao, Z., Rogozhnikov, A., Rollins, N., Wollenhaupt, P., et al. Zero-shot antibody design in a 24-well plate.bioRxiv, pp. 2025–07,
2025
-
[5]
Cho, Y ., Pacesa, M., Zhang, Z., Correia, B
doi: 10.1101/2025.01.08.631967. Cho, Y ., Pacesa, M., Zhang, Z., Correia, B. E., and Ovchin- nikov, S. Boltzdesign1: Inverting all-atom structure pre- diction model for generalized biomolecular binder de- sign.bioRxiv, pp. 2025–04,
-
[6]
URLhttps://www.biorxiv.org/content/ early/2023/05/25/2023.05.24.542194
doi: 10.1101/2023.05.24.542194. URLhttps://www.biorxiv.org/content/ early/2023/05/25/2023.05.24.542194. Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., ˇZ´ıdek, A., Bates, R., Blackwell, S., Yim, J., et al. Protein complex prediction with alphafold- multimer.biorxiv, pp. 2021–10,
-
[7]
Gong, C., Chen, X., Zhang, Y ., Song, Y ., Zhou, H., and Xiao, W. Protenix-mini: Efficient structure predictor via compact architecture, few-step diffusion and switchable plm.arXiv preprint arXiv:2507.11839,
-
[8]
Pacesa, M., Nickel, L., Schellhaas, C., Schmidt, J., Pyatova, E., Kissling, L., Barendse, P., Choudhury, J., Kapoor, S., Alcaraz-Serna, A., Cho, Y ., Ghamary, K. H., Vinu´e, L., Yachnin, B. J., Wollacott, A. M., Buckley, S., Westphal, A. H., Lindhoud, S., Georgeon, S., Goverde, C. A., Hatzopoulos, G. N., G¨onczy, P., Muller, Y . D., Schwank, G., Swarts, D...
-
[9]
Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction
doi: 10.1101/2025.06.14.659707. Qu, W., Ma, Y ., Ye, F., Lu, C., Zhou, Y ., Zhang, K., Wang, L., Gui, M., and Gu, Q. Seedproteo: Accurate de novo all-atom design of protein binders,
-
[10]
URLhttps: //arxiv.org/abs/2512.24192. Stark, H., Faltings, F., Choi, M., Xie, Y ., Hur, E., O’Donnell, T., Bushuiev, A., Uc ¸ar, T., Passaro, S., Mao, W., Reveiz, M., Bushuiev, R., Pluskal, T., Sivic, J., Kreis, K., Vahdat, A., Ray, S., Goldstein, J. T., Savinov, A., Hambalek, J. A., Gupta, A., Taquiri-Diaz, D. A., Zhang, Y ., Hatstat, A. K., Arada, A., K...
-
[11]
URLhttps://www.biorxiv.org/content/ early/2025/11/24/2025.11.20.689494
doi: 10.1101/2025.11.20.689494. URLhttps://www.biorxiv.org/content/ early/2025/11/24/2025.11.20.689494. Team, P., Ren, M., Sun, J., Guan, J., Liu, C., Gong, C., Wang, Y ., Wang, L., Cai, Q., Chen, X., and Xiao, W. Pxdesign: Fast, modular, and accu- rate de novo design of protein binders.bioRxiv,
-
[12]
URL https://www.biorxiv.org/content/ early/2025/08/16/2025.08.15.670450
doi: 10.1101/2025.08.15.670450. URL https://www.biorxiv.org/content/ early/2025/08/16/2025.08.15.670450. Van Kempen, M., Kim, S. S., Tumescheit, C., Mirdita, M., Lee, J., Gilchrist, C. L., S ¨oding, J., and Steinegger, M. Fast and accurate protein structure search with foldseek. Nature biotechnology, 42(2):243–246,
-
[13]
Zambaldi, V ., La, D., Chu, A. E., Patani, H., Danson, A. E., Kwan, T. O., Frerix, T., Schneider, R. G., Saxton, D., Thillaisundaram, A., et al. De novo design of high- affinity protein binders with alphaproteo.arXiv preprint arXiv:2409.08022,
-
[14]
URLhttps: //arxiv.org/abs/2510.22304. 11 ProtDBench: A Unified Benchmark of Protein Binder Design and Evaluation Target PDB ID Crop Hotspot Natural binder Binder length BHRF12wh6A2–158 A65, A74, A77, A82, A85, A93 BH3 helix 80–120 SC2RBD6m0jE333–526 E485, E489, E494, E500, E505 ACE2 receptor 80–120 IL-7RA3di3B17–209 B58, B80, B139 IL-7 50–120 PD-L15o45A17...
-
[15]
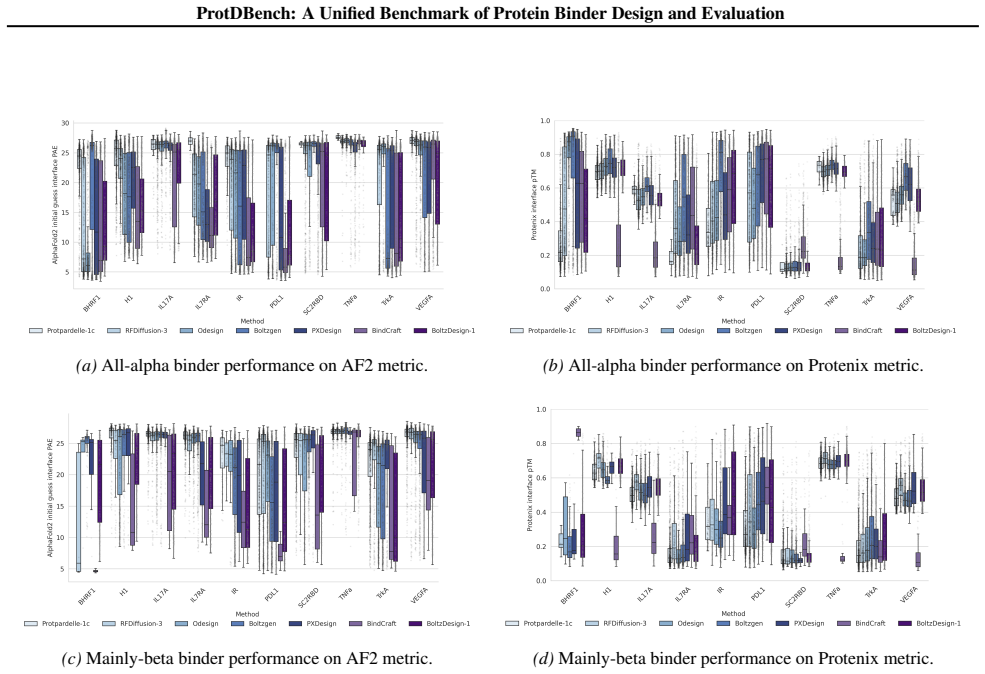
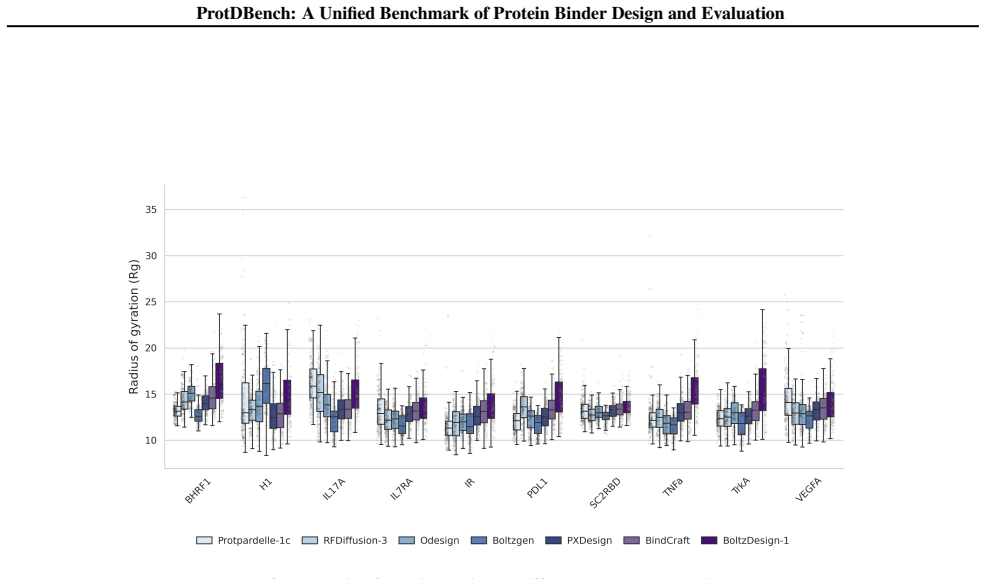
14 ProtDBench: A Unified Benchmark of Protein Binder Design and Evaluation (a)All-alpha binder performance on AF2 metric
With the exception of BindCraft, all methods were evaluated using their default code and parameters. 14 ProtDBench: A Unified Benchmark of Protein Binder Design and Evaluation (a)All-alpha binder performance on AF2 metric. (b)All-alpha binder performance on Protenix metric. (c)Mainly-beta binder performance on AF2 metric. (d)Mainly-beta binder performance...
2017
-
[16]
Multiple open-source variants of AF3 are released later, including Boltz-1 (Wohlwend et al., 2024), Boltz-2 (Passaro et al., 2025), Chai-1 (ChaiDiscovery et al.,
achieves higher pre- diction accuracy and is able to predict the joint structure of complexes including proteins, nucleic acids, small molecules, ions and modified residues. Multiple open-source variants of AF3 are released later, including Boltz-1 (Wohlwend et al., 2024), Boltz-2 (Passaro et al., 2025), Chai-1 (ChaiDiscovery et al.,
2024
-
[17]
These predictors are now integral to design and evaluation workflows
and Protenix (Chen et al., 2025). These predictors are now integral to design and evaluation workflows. 20
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.