Recognition: 3 theorem links
· Lean TheoremTree-Conditioned Edit Flows for Ancestral Sequence Reconstruction
Pith reviewed 2026-05-08 18:01 UTC · model grok-4.3
The pith
A tree-conditioned edit-flow model reconstructs ancestral sequences from descendants via paired bidirectional edit trajectories constrained to a common state.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper introduces a tree-conditioned edit-flow model for variable-length ASR. Given two descendant sequences and their branch distances to a shared ancestor, the model reconstructs the ancestor through paired bidirectional edit trajectories constrained to agree on a common ancestral state. On a benchmark of experimentally evolved sequences with only context-independent substitutions, the model does not match the accuracy of the best classical method, yet still achieves reasonable performance despite being trained on natural sequences that include insertions, deletions, and substitutions. On a benchmark of natural homologous sequences with abundant insertions and deletions, the model most
What carries the argument
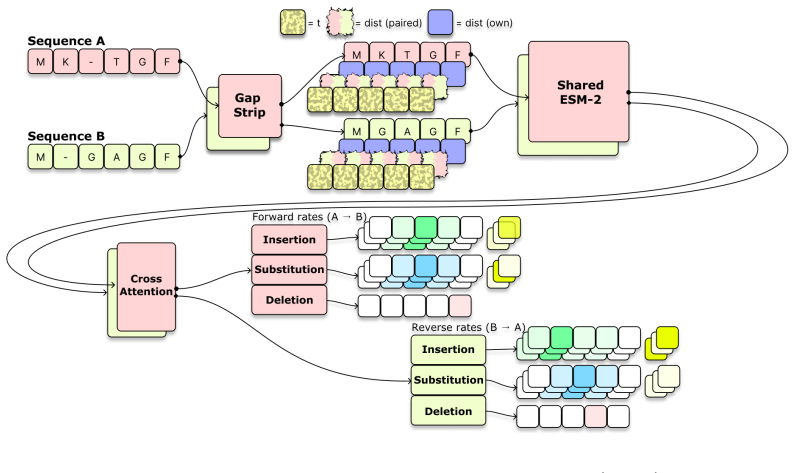
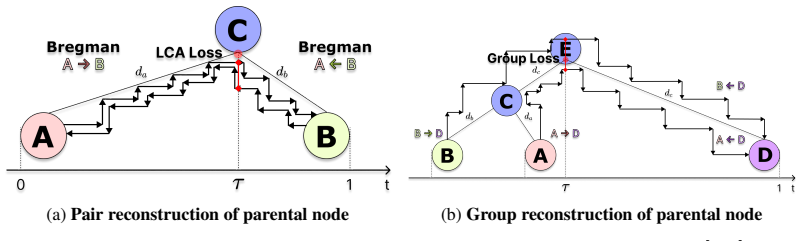
Tree-conditioned edit-flow model that reconstructs an ancestor by generating paired bidirectional edit trajectories from two descendants and enforcing agreement on a single ancestral state.
Load-bearing premise
Paired bidirectional edit trajectories trained on natural sequences will generalize to produce accurate ancestral reconstructions on both substitution-only and indel-rich cases when forced to agree on a common state.
What would settle it
An experimental evolution dataset where true ancestral sequences and all insertion, deletion, and substitution events are known in advance; the model would be falsified if its localized changes deviate from the recorded events more than classical methods do.
Figures
read the original abstract
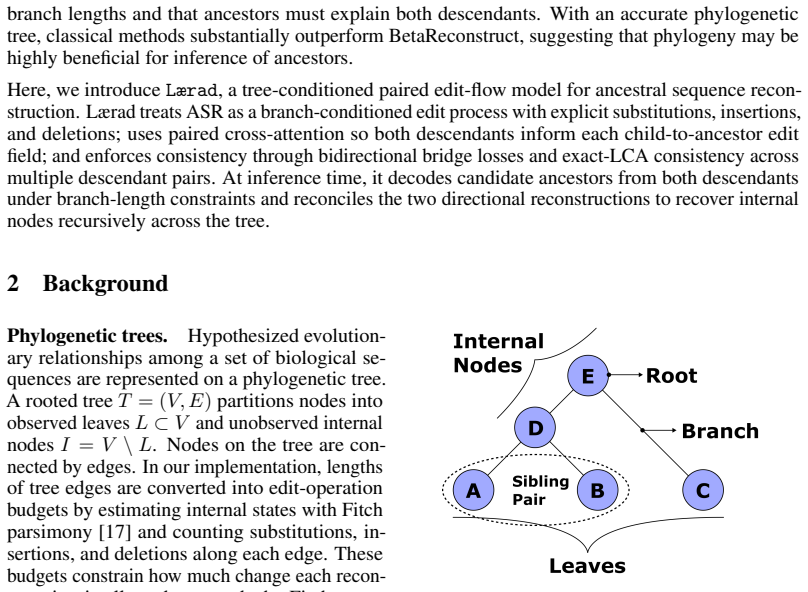
Ancestral sequence reconstruction (ASR) aims to infer extinct protein sequences at internal nodes of a phylogenetic tree. Classical ASR methods are typically based on continuous-time Markov substitution models, but they treat sites largely independently and handle insertions and deletions only weakly or not at all. We introduce a tree-conditioned edit-flow model for variable-length ASR. Given two descendant sequences and their branch distances to a shared ancestor, the model reconstructs the ancestor through paired bidirectional edit trajectories constrained to agree on a common ancestral state. On a benchmark of experimentally evolved sequences with only context-independent substitutions, the model does not match the accuracy of the best classical method, yet still achieves reasonable performance despite being trained on natural sequences that include insertions, deletions, and substitutions. On a benchmark of natural homologous sequences with abundant insertions and deletions, the model most accurately localizes inferred evolutionary change.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a tree-conditioned edit-flow model for ancestral sequence reconstruction (ASR). Given two descendant sequences and their branch distances to a shared ancestor, the model reconstructs the ancestor via paired bidirectional edit trajectories constrained to agree on a common ancestral state. This is positioned as an advance over classical continuous-time Markov substitution models, which treat sites independently and handle indels weakly. The model is trained on natural sequences (including indels and substitutions) and evaluated on two benchmarks: experimentally evolved sequences with only context-independent substitutions (where it achieves reasonable but sub-optimal accuracy relative to the best classical method) and natural homologous sequences with abundant indels (where it most accurately localizes inferred evolutionary change).
Significance. If the central claims hold, the work offers a novel data-driven framework for variable-length ASR that directly incorporates indels via edit flows, addressing a clear limitation of traditional substitution models. Training on natural data and the bidirectional agreement constraint are strengths that could enable better handling of complex evolutionary histories. However, the underperformance on substitution-only data with available ground truth and the reliance on relative localization against inferred changes (without independent ground truth) temper the significance; the approach may still prove useful if its generalization properties can be more rigorously established.
major comments (2)
- [Abstract] Abstract: the claim that the model 'most accurately localizes inferred evolutionary change' on natural homologous sequences with abundant indels rests on accuracy measured against other inference procedures; without independent ground truth for the true ancestral states, it is unclear whether the bidirectional edit trajectories recover the actual history or merely a consistent but incorrect one that agrees with the baselines.
- [Abstract] Abstract: on the benchmark of experimentally evolved sequences with only substitutions, the model underperforms the best classical method despite the availability of ground truth; this indicates that the edit distribution learned from natural data (which includes indels) does not automatically align with the true substitution process, raising concerns about the generalization assumption for both benchmarks.
minor comments (1)
- [Abstract] Abstract: comparative performance numbers are reported without accompanying details on model architecture, training procedure, statistical tests, error bars, or precise definitions of the localization metric.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed feedback on our manuscript. We address each major comment below in a point-by-point manner. We agree with the concerns about qualifying claims in the absence of ground truth and have revised the abstract and added discussion text to more precisely describe the evaluation and its limitations.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the model 'most accurately localizes inferred evolutionary change' on natural homologous sequences with abundant indels rests on accuracy measured against other inference procedures; without independent ground truth for the true ancestral states, it is unclear whether the bidirectional edit trajectories recover the actual history or merely a consistent but incorrect one that agrees with the baselines.
Authors: We agree that the lack of independent ground truth for natural sequences means we cannot definitively claim recovery of the true history. The original abstract already qualifies the result as localizing 'inferred evolutionary change' to reflect comparison against other methods. To address the possibility of consistent but incorrect agreement, we have revised the abstract to explicitly note that results are relative to baseline inferences and added a new paragraph in the Discussion section acknowledging this limitation while noting that agreement across independent methods serves as a standard proxy in indel-rich ASR settings. revision: yes
-
Referee: [Abstract] Abstract: on the benchmark of experimentally evolved sequences with only substitutions, the model underperforms the best classical method despite the availability of ground truth; this indicates that the edit distribution learned from natural data (which includes indels) does not automatically align with the true substitution process, raising concerns about the generalization assumption for both benchmarks.
Authors: The referee correctly identifies that our model underperforms the strongest classical substitution model on the experimental benchmark with ground truth. We report this result transparently and do not claim superiority for pure substitution cases. The manuscript instead highlights reasonable accuracy despite training on natural data containing indels. We have revised the abstract to state this limitation more explicitly and expanded the Results and Discussion sections to discuss the implications for generalization, noting that the model's primary intended use is for variable-length sequences with mixed indels and substitutions where classical models are weaker. revision: yes
Circularity Check
No significant circularity; model trained on external data
full rationale
The paper introduces a tree-conditioned edit-flow model trained on natural homologous sequences to perform ancestral sequence reconstruction. No equations, derivations, or self-referential definitions appear in the provided abstract or description. The central claims rely on empirical training from external data and comparisons to classical methods on benchmarks, without any load-bearing steps that reduce predictions to fitted inputs by construction or self-citation chains. This is a standard non-circular empirical modeling approach.
Axiom & Free-Parameter Ledger
invented entities (1)
-
tree-conditioned edit-flow model
no independent evidence
Lean theorems connected to this paper
-
IndisputableMonolith/Cost (Jcost, FunctionalEquation)washburn_uniqueness_aczel unclearFollowing Edit Flows [24], the base term trains a continuous-time edit-rate field by penalizing total predicted edit mass while rewarding rates assigned to edits that move a sampled bridge state toward the target endpoint.
Reference graph
Works this paper leans on
-
[1]
Linus Pauling, Emile Zuckerkandl, Thormod Henriksen, and Rolf Lövstad. Chemical paleogenetics. molecular "restoration studies" of extinct forms of life.Acta Chemica Scandinavica, 17 supl.:9–16, 1963. ISSN 0904-213X. doi: 10.3891/acta.chem.scand.17s-0009
-
[2]
A. G. A. Selberg, E. A. Gaucher, and D. A. Liberles. Ancestral sequence reconstruction: From chemical paleogenetics to maximum likelihood algorithms and beyond.J Mol Evol, 89(3):157–164, 2021. ISSN 1432-1432 (Electronic) 0022-2844 (Print) 0022-2844 (Linking). doi: 10.1007/s00239-021-09993-1. URL https://www.ncbi.nlm.nih.gov/pubmed/33486547
-
[3]
K. Prakinee, S. Phaisan, S. Kongjaroon, and P. Chaiyen. Ancestral sequence reconstruction for designing biocatalysts and investigating their functional mechanisms.JACS Au, 4(12):4571–4591, 2024. ISSN 2691-3704 (Electronic) 2691-3704 (Linking). doi: 10.1021/jacsau.4c00653. URL https://www.ncbi. nlm.nih.gov/pubmed/39735918
-
[4]
M. A. Spence, J. A. Kaczmarski, J. W. Saunders, and C. J. Jackson. Ancestral sequence reconstruction for protein engineers.Curr Opin Struct Biol, 69:131–141, 2021. ISSN 1879-033X (Electronic) 0959- 440X (Linking). doi: 10.1016/j.sbi.2021.04.001. URL https://www.ncbi.nlm.nih.gov/pubmed/ 34023793
-
[5]
J. M. Koshi and R. A. Goldstein. Probabilistic reconstruction of ancestral protein sequences.J Mol Evol, 42(2):313–20, 1996. ISSN 0022-2844 (Print) 0022-2844 (Linking). doi: 10.1007/BF02198858. URL https://www.ncbi.nlm.nih.gov/pubmed/8919883
- [6]
-
[7]
John P. Huelsenbeck and Jonathan P. Bollback. Empirical and hierarchical bayesian estimation of ancestral states.Systematic Biology, 50(3):351–366, 2001. doi: 10.1080/10635150119871
-
[8]
C. Norn and I. Andre. Atomistic simulation of protein evolution reveals sequence covariation and time- dependent fluctuations of site-specific substitution rates.PLoS Comput Biol, 19(3):e1010262, 2023. ISSN 1553-7358 (Electronic) 1553-734X (Print) 1553-734X (Linking). doi: 10.1371/journal.pcbi.1010262. URL https://www.ncbi.nlm.nih.gov/pubmed/36961827
-
[9]
D. D. Pollock, G. Thiltgen, and R. A. Goldstein. Amino acid coevolution induces an evolutionary Stokes shift.Proc Natl Acad Sci U S A, 109(21):E1352–9, 2012. ISSN 1091-6490 (Electronic) 0027-8424 (Print) 0027-8424 (Linking). doi: 10.1073/pnas.1120084109. URL https://www.ncbi.nlm.nih.gov/ pubmed/22547823
-
[10]
Joseph Felsenstein. Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters.Systematic Biology, 22(3):240–249, 09 1973. ISSN 1063-5157. doi: 10.1093/sysbio/22.3.240. URLhttps://doi.org/10.1093/sysbio/22.3.240
-
[11]
Pattern Recognition 127 (2022), 108611
S. Savino, T. Desmet, and J. Franceus. Insertions and deletions in protein evolution and engineering. Biotechnol Adv, 60:108010, 2022. ISSN 1873-1899 (Electronic) 0734-9750 (Linking). doi: 10.1016/j. biotechadv.2022.108010. URLhttps://www.ncbi.nlm.nih.gov/pubmed/35738511
work page doi:10.1016/j 2022
-
[12]
A. Toth-Petroczy and D. S. Tawfik. Protein insertions and deletions enabled by neutral roaming in sequence space.Mol Biol Evol, 30(4):761–71, 2013. ISSN 1537-1719 (Electronic) 0737-4038 (Linking). doi: 10.1093/molbev/mst003. URLhttps://www.ncbi.nlm.nih.gov/pubmed/23315956
-
[13]
F. Hormozdiari, R. Salari, M. Hsing, A. Schonhuth, S. K. Chan, S. C. Sahinalp, and A. Cherkasov. The effect of insertions and deletions on wirings in protein-protein interaction networks: a large-scale study.J Comput Biol, 16(2):159–67, 2009. ISSN 1557-8666 (Electronic) 1066-5277 (Linking). doi: 10.1089/cmb.2008.03TT. URLhttps://www.ncbi.nlm.nih.gov/pubme...
-
[14]
Gholamhossein Jowkar, J¯ulija Peˇcerska, Massimo Maiolo, Manuel Gil, and Maria Anisimova. ARPIP: Ancestral sequence reconstruction with insertions and deletions under the Poisson indel process.Systematic Biology, 72(2):307–318, 07 2022. ISSN 1063-5157. doi: 10.1093/sysbio/syac050. URL https://doi. org/10.1093/sysbio/syac050
-
[15]
Reconstruction of ancestral protein sequences using autoregressive generative models.Molecular Biology and Evolution, 42(4):msaf070, 04
Matteo De Leonardis, Andrea Pagnani, and Pierre Barrat-Charlaix. Reconstruction of ancestral protein sequences using autoregressive generative models.Molecular Biology and Evolution, 42(4):msaf070, 04
-
[16]
ISSN 1537-1719. doi: 10.1093/molbev/msaf070. URL https://doi.org/10.1093/molbev/ msaf070. 10
-
[17]
Ancestral sequence reconstruction using generative models.bioRxiv, 2026
Edo Dotan, Elya Wygoda, Asaf Schers, Iris Lyubman, Yonatan Belinkov, and Tal Pupko. Ancestral sequence reconstruction using generative models.bioRxiv, 2026. doi: 10.64898/2026.01.18.700141. URL https://www.biorxiv.org/content/early/2026/01/21/2026.01.18.700141
-
[18]
Walter M. Fitch. Toward defining the course of evolution: Minimum change for a specific tree topology. Systematic Zoology, 20(4):406–416, 1971. ISSN 00397989. URL http://www.jstor.org/stable/ 2412116
1971
-
[19]
David T. Jones, William R. Taylor, and Janet M. Thornton. The rapid generation of mutation data matrices from protein sequences.Bioinformatics, 8(3):275–282, 06 1992. ISSN 1367-4803. doi: 10.1093/ bioinformatics/8.3.275. URLhttps://doi.org/10.1093/bioinformatics/8.3.275
-
[20]
Simon Whelan and Nick Goldman. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach.Molecular Biology and Evolution, 18(5):691–699, 05 2001. ISSN 0737-4038. doi: 10.1093/oxfordjournals.molbev.a003851. URL https://doi.org/10. 1093/oxfordjournals.molbev.a003851
-
[21]
Si Quang Le and Olivier Gascuel. An improved general amino acid replacement matrix.Molecular Biology and Evolution, 25(7):1307–1320, 07 2008. ISSN 0737-4038. doi: 10.1093/molbev/msn067. URL https://doi.org/10.1093/molbev/msn067
-
[22]
Z Yang. Maximum-likelihood estimation of phylogeny from dna sequences when substitution rates differ over sites.Molecular Biology and Evolution, 10(6):1396–1401, 11 1993. ISSN 0737-4038. doi: 10.1093/oxfordjournals.molbev.a040082. URL https://doi.org/10.1093/oxfordjournals. molbev.a040082
-
[23]
Tal Pupko, Itsik Pe’er, Ron Shamir, and Dan Graur. A fast algorithm for joint reconstruction of ancestral amino acid sequences.Molecular Biology and Evolution, 17(6):890–896, 06 2000. ISSN 0737-4038. doi: 10.1093/oxfordjournals.molbev.a026369. URL https://doi.org/10.1093/oxfordjournals. molbev.a026369
-
[24]
Andrew Campbell, Jason Yim, Regina Barzilay, Tom Rainforth, and Tommi Jaakkola. Generative flows on discrete state-spaces: Enabling multimodal flows with applications to protein co-design, 2024. URL https://arxiv.org/abs/2402.04997
- [25]
-
[26]
Language models of protein sequences at the scale of evolution enable accurate structure prediction.Science, 379(6637):1123–1130,
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction.Science, 379(6637):1123–1130,
-
[27]
doi: 10.1126/science.ade2574
-
[28]
EvoFlows: Evolutionary Edit-Based Flow-Matching for Protein Engineering
Nicolas Deutschmann, Constance Ferragu, Jonathan D. Ziegler, Shayan Aziznejad, and Eli Bixby. EvoFlows: Evolutionary edit-based flow-matching for protein engineering, 2026. URL https://arxiv. org/abs/2603.11703
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[29]
T.J. Lambert. FPbase: a community-editable fluorescent protein database.Nature Methods, 16,
-
[30]
URL https://www.nature.com/articles/ s41592-019-0352-8
doi: https://doi.org/10.1038/s41592-019-0352-8. URL https://www.nature.com/articles/ s41592-019-0352-8
-
[31]
Jaime Huerta-Cepas, Damian Szklarczyk, Davide Heller, Ana Hernández-Plaza, Sofia K Forslund, Helen Cook, Daniel R Mende, Ivica Letunic, Thomas Rattei, Lars J Jensen, Christian von Mering, and Peer Bork. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses.Nucleic Acids Research...
-
[32]
Rohan Maddamsetti, Daniel T. Johnson, Stephanie J. Spielman, Katherine L. Petrie, Debora S. Marks, and Justin R. Meyer. Gain-of-function experiments with bacteriophage lambda uncover residues under diversifying selection in nature.Evolution, 72(10):2234–2243, 10 2018. ISSN 0014-3820. doi: 10.1111/ evo.13586. URLhttps://doi.org/10.1111/evo.13586
-
[33]
Ryan N. Randall, Caelan E. Radford, Kelsey A. Roof, Divya K. Natarajan, and Eric A. Gaucher. An exper- imental phylogeny to benchmark ancestral sequence reconstruction.Nature Communications, 7, 2016. doi: https://doi.org/10.1038/ncomms12847. URLhttps://www.nature.com/articles/ncomms12847
-
[34]
Stéphane Guindon, Jean-François Dufayard, Vincent Lefort, Maria Anisimova, Wim Hordijk, and Olivier Gascuel. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0.Systematic Biology, 59(3):307–321, 2010. doi: 10.1093/sysbio/syq010. 11
-
[35]
BioBERT: a pre-trained biomedical language representation model for biomedical text mining,
A. Oliva, S. Pulicani, V . Lefort, L. Brehelin, O. Gascuel, and S. Guindon. Accounting for ambiguity in ancestral sequence reconstruction.Bioinformatics, 35(21):4290–4297, 2019. doi: 10.1093/bioinformatics/ btz249
-
[36]
Schmidt, Arndt von Haeseler, and Bui Quang Minh
Lam-Tung Nguyen, Heiko A. Schmidt, Arndt von Haeseler, and Bui Quang Minh. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies.Molecular Biology and Evolution, 32(1):268–274, 2015. doi: 10.1093/molbev/msu300
-
[37]
MMseqs 2 enables sensitive protein sequence searching for the analysis of massive data sets
Martin Steinegger and Johannes Söding. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets.Nature Biotechnology, 2017. doi: 10.1038/nbt.3988
-
[38]
Morgan N. Price, Paramvir S. Dehal, and Adam P. Arkin. Fasttree 2: Approximately maximum-likelihood trees for large alignments.PLOS ONE, 5(3):e9490, 2010. doi: 10.1371/journal.pone.0009490
-
[39]
Kazutaka Katoh and Daron M. Standley. Mafft multiple sequence alignment software version 7: Im- provements in performance and usability.Molecular Biology and Evolution, 30(4):772–780, 2013. doi: 10.1093/molbev/mst010
-
[40]
Alexey M Kozlov, Diego Darriba, Tomáš Flouri, Benoit Morel, and Alexandros Stamatakis. Raxml-ng: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference.Bioinformatics, 35(21):4453–4455, 11 2019. ISSN 1367-4803. doi: 10.1093/bioinformatics/btz305. URL https: //doi.org/10.1093/bioinformatics/btz305
-
[41]
Ziheng Yang. PAML 4: Phylogenetic analysis by maximum likelihood.Molecular Biology and Evolution, 24(8):1586–1591, 2007. doi: 10.1093/molbev/msm088. 12 A Technical appendices and supplementary material A.1 ASR Classical ASR likelihood with among-site rate variation.Classical protein ASR models evolution at each aligned site as a continuous-time Markov cha...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.