Recognition: 2 theorem links
· Lean TheoremWhen Does Gene Regulatory Network Inference Break? A Controlled Diagnostic Study of Causal and Correlational Methods on Single-Cell Data
Pith reviewed 2026-05-08 18:06 UTC · model grok-4.3
The pith
Causal methods for gene regulatory network inference from single-cell data outperform correlation baselines only in clean regimes without dropout or latent confounders.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
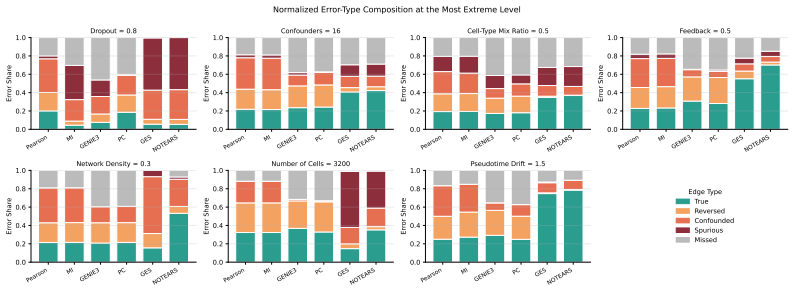
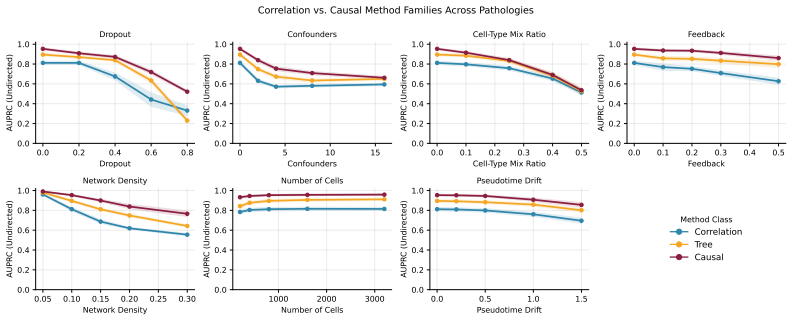
Across 6,120 controlled experiments isolating seven pathologies, causal methods dominate in clean and structurally favorable regimes, but specific pathologies (notably dropout and latent confounders) selectively neutralize their advantages. Methods with similar aggregate accuracy commit qualitatively different errors. Joint effects of multiple pathologies are sub-additive while also exposing density-conditional cross-overs invisible to single-dial analysis.
What carries the argument
A controlled diagnostic framework that independently varies seven biologically motivated pathologies in simulated single-cell RNA-seq data to track degradation of six representative inference methods.
If this is right
- Causal methods are preferable only when dropout and latent confounders can be ruled out or corrected in the data.
- Error-type decomposition distinguishes methods even when their overall accuracy scores are similar.
- Joint effects of pathologies being sub-additive means fixing the strongest single problem can produce larger gains than expected.
- Network density changes how pathologies interact, so it must be tracked in any comparative evaluation.
Where Pith is reading between the lines
- Practitioners should first check dropout levels and potential confounders in their single-cell data before defaulting to causal methods.
- Method developers could target robustness to dropout and latent variables to make causal advantages usable in typical datasets.
- The same isolation approach could be applied to other biological inference tasks to reveal regime-specific method strengths.
Load-bearing premise
The simulation model with its seven isolated pathologies captures the dominant failure modes in real single-cell RNA-seq data.
What would settle it
Apply the same methods to real single-cell datasets in which dropout rates and latent confounders have been independently measured or experimentally controlled and check whether performance rankings match the simulation patterns.
Figures








read the original abstract
Despite theoretical advantages, causal methods for Gene Regulatory Network (GRN) inference from single-cell RNA-seq data consistently fail to match or outperform correlation-based baselines in many realistic benchmarks, a persistent puzzle which casts doubt on the value of causality for this task. We argue that existing benchmarks are insufficiently controlled to answer this question because they evaluate on real or semi-real data where multiple pathologies co-occur, confounding failure modes, and obscuring the specific conditions under which different inference methods excel or fail. To address this gap, we introduce a controlled diagnostic framework that isolates seven biologically motivated pathologies (dropout, latent confounders, cell-type mixing, feedback loops, network density, sample size, and pseudotime drift) and measure how six representative methods spanning three inference paradigms degrade as each pathology intensifies. Across 6,120 controlled experiments, we find that causal methods genuinely dominate in clean and structurally favorable regimes, but specific pathologies (notably dropout and latent confounders) selectively neutralize their advantages. We further introduce an error-type decomposition that reveals methods with similar aggregate accuracy commit qualitatively different errors. To probe whether single-pathology effects persist when multiple stressors co-occur, we perform an interaction sweep over the three most impactful pathologies and find that their joint effects are sub-additive, while also exposing density-conditional cross-overs invisible to single-dial analysis. Our findings offer a nuanced understanding of when and why different methods succeed or fail for GRN inference, providing actionable insights for method development and practical guidance for practitioners.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a controlled simulation framework that isolates seven pathologies (dropout, latent confounders, cell-type mixing, feedback loops, network density, sample size, pseudotime drift) in single-cell RNA-seq data for GRN inference. Across 6,120 experiments on six methods spanning causal, correlational, and other paradigms, it reports that causal methods outperform baselines in clean regimes but have their advantages selectively neutralized by dropout and latent confounders; an error decomposition shows qualitatively different failure modes, and an interaction sweep reveals sub-additive joint effects with density-conditional cross-overs.
Significance. If the simulations faithfully reproduce real scRNA-seq statistics, the scale of the controlled experiments, the error-type decomposition, and the interaction analysis provide actionable diagnostics for when causal GRN methods are likely to succeed or fail, offering clearer guidance than existing mixed-pathology benchmarks. The sub-additive interaction results and the demonstration that aggregate accuracy can mask distinct error profiles are particularly valuable contributions.
major comments (1)
- [Section 3] Simulation framework (Section 3): the generative model for the seven pathologies (e.g., zero-inflation implementation for dropout, injection of latent confounders into regulatory dynamics) is not calibrated or validated against empirical moments (mean-variance relationships, zero fractions, or known GRN topologies) from real single-cell datasets of the same cell types. This is load-bearing because the central claims about selective neutralization and sub-additive interactions rest on the pathologies being realistic rather than simulation artifacts.
minor comments (3)
- [Abstract] Abstract: the phrase 'three inference paradigms' is not expanded; explicitly naming the paradigms (causal, correlational, and the third) would improve clarity for readers unfamiliar with the GRN literature.
- [Results figures] Figure captions (e.g., those summarizing the 6,120-experiment results): some panels lack explicit axis labels for pathology intensity levels, making it harder to map quantitative degradation curves to the seven isolated factors.
- [Methods] The error decomposition is introduced without a formal definition or pseudocode; adding a short algorithmic description would help readers reproduce the qualitative error-type distinctions.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback. The single major comment is addressed point-by-point below. We agree that additional calibration and validation steps will strengthen the paper and commit to incorporating them in the revision.
read point-by-point responses
-
Referee: [Section 3] Simulation framework (Section 3): the generative model for the seven pathologies (e.g., zero-inflation implementation for dropout, injection of latent confounders into regulatory dynamics) is not calibrated or validated against empirical moments (mean-variance relationships, zero fractions, or known GRN topologies) from real single-cell datasets of the same cell types. This is load-bearing because the central claims about selective neutralization and sub-additive interactions rest on the pathologies being realistic rather than simulation artifacts.
Authors: We agree that the simulation framework would benefit from explicit calibration and validation against real data. While the primary goal of the study is controlled isolation of individual pathologies (rather than faithful replication of any specific real dataset), we will revise Section 3 to add: (1) direct comparisons of key simulated statistics (mean-variance relationships, zero fractions, and marginal distributions) to empirical moments drawn from representative real scRNA-seq datasets of common cell types; (2) a brief discussion of how the chosen ranges for each pathology parameter align with values reported in the literature; and (3) a short sensitivity analysis showing that the reported qualitative findings (selective neutralization by dropout and latent confounders, sub-additive interactions) remain stable under modest perturbations of the generative parameters. These additions will make the realism of the controlled experiments more transparent without altering the core experimental design or conclusions. revision: yes
Circularity Check
No circularity: purely empirical simulation study with no derivations
full rationale
This paper conducts a controlled diagnostic study by running 6,120 simulation experiments that isolate seven pathologies and measure degradation in six GRN inference methods. There are no equations, fitted parameters renamed as predictions, self-citation chains, or ansatzes that reduce the central claims to their own inputs. All findings (causal dominance in clean regimes, selective neutralization by dropout and confounders, sub-additive interactions) are direct empirical measurements, making the work self-contained.
Axiom & Free-Parameter Ledger
free parameters (1)
- pathology intensity parameters
axioms (1)
- domain assumption The simulation model with independently controllable pathologies accurately reflects the dominant failure modes of real single-cell RNA-seq data
Lean theorems connected to this paper
-
Cost.FunctionalEquation / Foundation.AlphaCoordinateFixation: RS forces J(x)=½(x+x⁻¹)−1 via ratio symmetry; paper uses an unrelated linear additive-noise SCM with no cost-function content.washburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
X^(c)_j = sum_{i in PG(j)} W_ij X^(c)_i + epsilon^(c)_j, epsilon iid ~ N(0, sigma^2)
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Zheng, Xun and Aragam, Bryon and Ravikumar, Pradeep K and Xing, Eric P , booktitle=
-
[2]
Bello, Kevin and Aragam, Bryon and Ravikumar, Pradeep , booktitle=
-
[3]
2000 , publisher=
Causation, Prediction, and Search , author=. 2000 , publisher=
2000
-
[4]
Journal of Machine Learning Research , volume=
Optimal Structure Identification with Greedy Search , author=. Journal of Machine Learning Research , volume=
-
[5]
2009 , publisher=
Causality: Models, Reasoning, and Inference , author=. 2009 , publisher=
2009
-
[6]
2019 , organization=
Yu, Yue and Chen, Jie and Gao, Tian and Yu, Mo , booktitle=. 2019 , organization=
2019
-
[7]
Gradient-Based Neural
Lachapelle, S. Gradient-Based Neural. International Conference on Learning Representations , year=
-
[8]
Advances in Neural Information Processing Systems , volume=
Differentiable Causal Discovery from Interventional Data , author=. Advances in Neural Information Processing Systems , volume=
-
[9]
PLoS ONE , volume=
Inferring Regulatory Networks from Expression Data Using Tree-Based Methods , author=. PLoS ONE , volume=. 2010 , publisher=
2010
-
[10]
Bioinformatics , volume=
Moerman, Thomas and Aibar Santos, Sara and Bravo Gonz. Bioinformatics , volume=. 2019 , publisher=
2019
-
[11]
Nature Methods , volume=
Aibar, Sara and Bravo Gonz. Nature Methods , volume=. 2017 , publisher=
2017
-
[12]
Nature Methods , volume=
Bravo Gonz. Nature Methods , volume=. 2023 , publisher=
2023
-
[13]
Nature Computational Science , volume=
Modeling Gene Regulatory Networks Using Neural Network Architectures , author=. Nature Computational Science , volume=. 2021 , publisher=
2021
-
[14]
2024 , publisher=
Cui, Haotian and Wang, Chloe and Maan, Hassaan and Pang, Kuan and Luo, Fengning and Duan, Nan and Wang, Bo , journal=. 2024 , publisher=
2024
-
[15]
Nature , volume=
Transfer Learning Enables Predictions in Network Biology , author=. Nature , volume=. 2023 , publisher=
2023
-
[16]
2020 , publisher=
Dibaeinia, Payam and Sinha, Saurabh , journal=. 2020 , publisher=
2020
-
[17]
Nature Methods , volume=
Benchmarking Algorithms for Gene Regulatory Network Inference from Single-Cell Transcriptomic Data , author=. Nature Methods , volume=. 2020 , publisher=
2020
-
[18]
PLoS ONE , volume=
Towards a Rigorous Assessment of Systems Biology Models: The DREAM3 Challenges , author=. PLoS ONE , volume=. 2010 , publisher=
2010
-
[19]
Nature Methods , volume=
Wisdom of Crowds for Robust Gene Network Inference , author=. Nature Methods , volume=. 2012 , publisher=
2012
-
[20]
2025 , publisher=
Chevalley, Mathieu and Roohani, Yusuf H and Mehrjou, Arash and Leskovec, Jure and Schwab, Patrick , journal=. 2025 , publisher=
2025
-
[21]
2025 , doi=
Nourisa, Jalil and Passemiers, Antoine and Stock, Marco and Zeller-Plumhoff, Berit and Cannoodt, Robrecht and Arnold, Christian and Tong, Alexander and Hartford, Jason and Scialdone, Antonio and Moreau, Yves and Li, Yang and Luecken, Malte D , journal=. 2025 , doi=
2025
-
[22]
Mapping Information-Rich Genotype--Phenotype Landscapes with Genome-Scale
Replogle, Joseph M and Saunders, Reuben A and Pogson, Angela N and Hussmann, Jeffrey A and Lenail, Alexander and Guna, Alina and Mascibroda, Lauren and Wagner, Eric J and Adelman, Karen and Lithwick-Yanai, Gila and others , journal=. Mapping Information-Rich Genotype--Phenotype Landscapes with Genome-Scale. 2022 , publisher=
2022
-
[23]
Predicting Transcriptional Outcomes of Novel Multigene Perturbations with
Roohani, Yusuf and Huang, Kexin and Leskovec, Jure , journal=. Predicting Transcriptional Outcomes of Novel Multigene Perturbations with. 2024 , publisher=
2024
-
[24]
Nature Methods , volume=
Deep-Learning-Based Gene Perturbation Effect Prediction Does Not Yet Outperform Simple Linear Baselines , author=. Nature Methods , volume=. 2025 , publisher=
2025
-
[25]
Advances in Neural Information Processing Systems , volume=
Large-Scale Differentiable Causal Discovery of Factor Graphs , author=. Advances in Neural Information Processing Systems , volume=
-
[26]
1991 , publisher=
Elements of Information Theory , author=. 1991 , publisher=
1991
-
[27]
Nature , volume=
Dissecting Cell Identity via Network Inference and In Silico Gene Perturbation , author=. Nature , volume=. 2023 , publisher=
2023
-
[28]
Proceedings of the 41st International Conference on Machine Learning , pages=
Stable Differentiable Causal Discovery , author=. Proceedings of the 41st International Conference on Machine Learning , pages=
-
[29]
Nature Communications , volume=
Kalfon, J. Nature Communications , volume=. 2025 , publisher=
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.