Trans-dimensional Bayesian model averaging for ¹³C-based metabolic flux analysis: Evidence-based flux inference under structural model uncertainty
Pith reviewed 2026-06-29 22:58 UTC · model grok-4.3
The pith
Bayesian model set averaging produces robust flux estimates by averaging over many possible metabolic network structures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Our approach combines reversible jump Markov chain Monte Carlo for trans-dimensional exploration of model spaces with diffusive nested sampling for robust estimation of model evidences, enabling averaging over large families of metabolic network models.
What carries the argument
Bayesian model set averaging, which uses reversible jump Markov chain Monte Carlo for trans-dimensional model exploration and diffusive nested sampling for model evidence estimation to average fluxes over large families of network structures.
If this is right
- Flux estimates remain stable when multiple network configurations fit the isotope data equally well.
- The method identifies cases where competing models cannot be statistically distinguished.
- Increasing data informativeness improves recovery of both supported model structures and flux values.
- The framework provides a practical way to handle misspecification in metabolic network models for 13C-MFA.
Where Pith is reading between the lines
- The scalability claim suggests the method could guide experimental design by quantifying how much labeling data is needed to resolve network ambiguities.
- Extension to genome-scale networks would require checking whether the samplers maintain mixing when the model space grows further.
- Integration with additional data types such as proteomics could further reduce the effective number of competing models.
Load-bearing premise
Reversible jump MCMC and diffusive nested sampling can practically explore and give reliable evidence estimates for model spaces containing billions of variants.
What would settle it
A synthetic data set generated from a known true network where the method fails to assign high posterior weight to the generating model or produces inaccurate averaged fluxes despite data sufficient to distinguish structures.
Figures

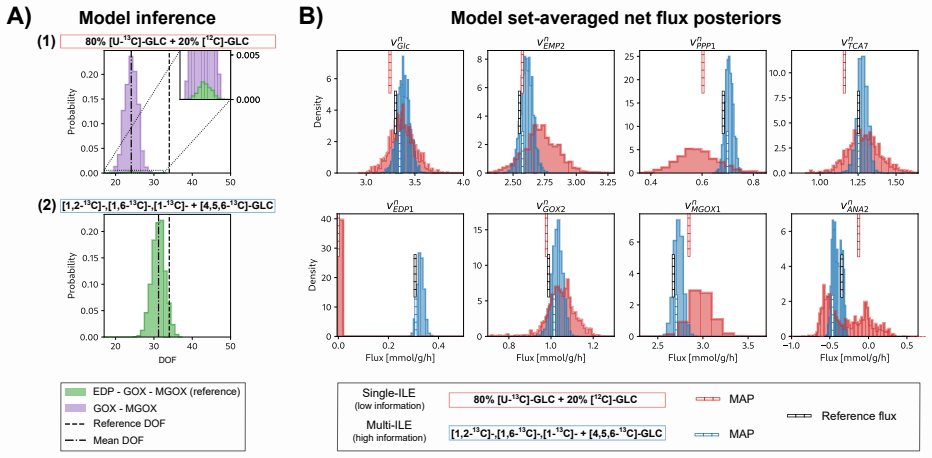
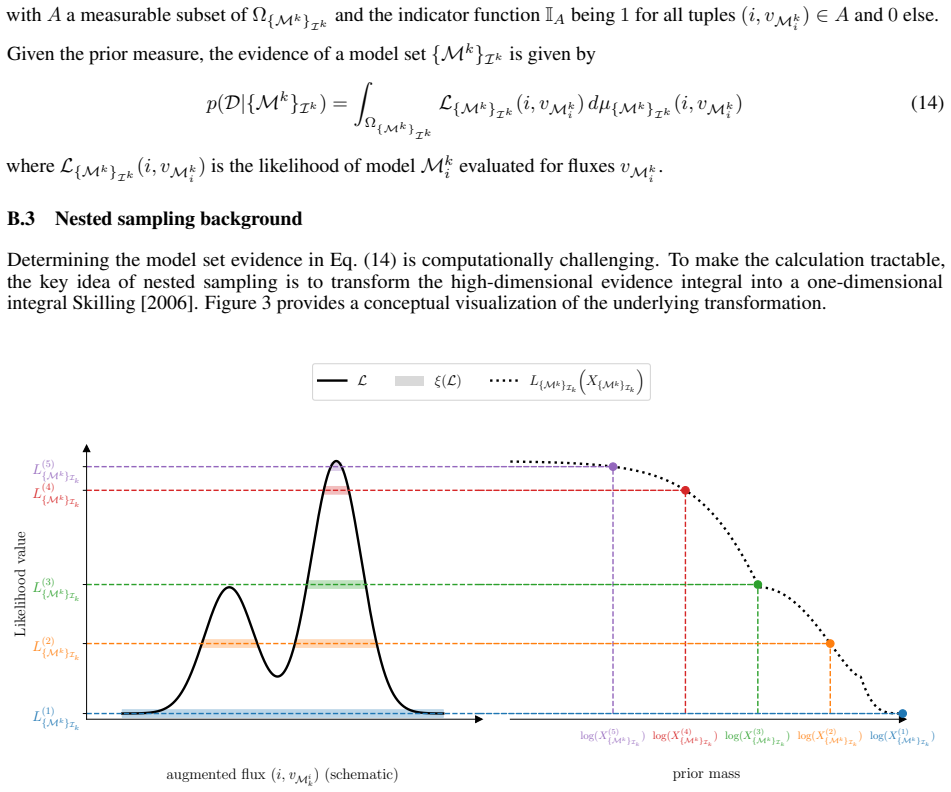
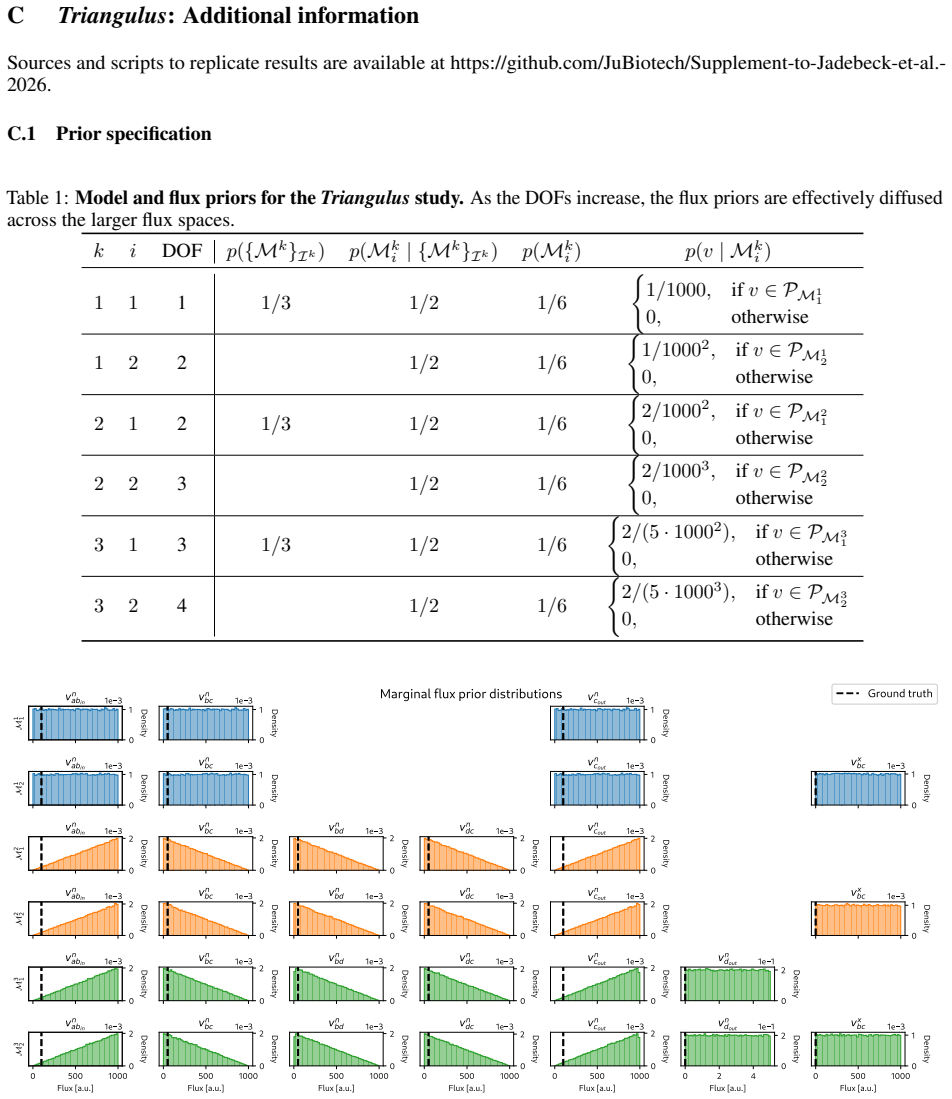
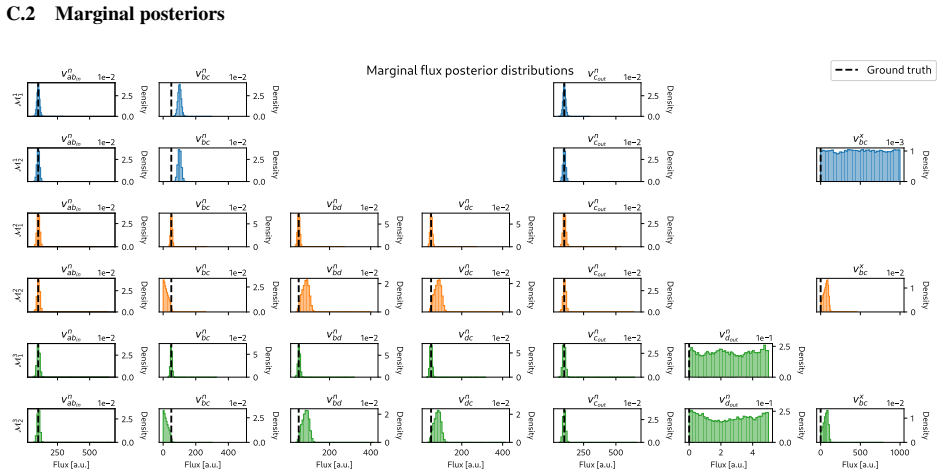
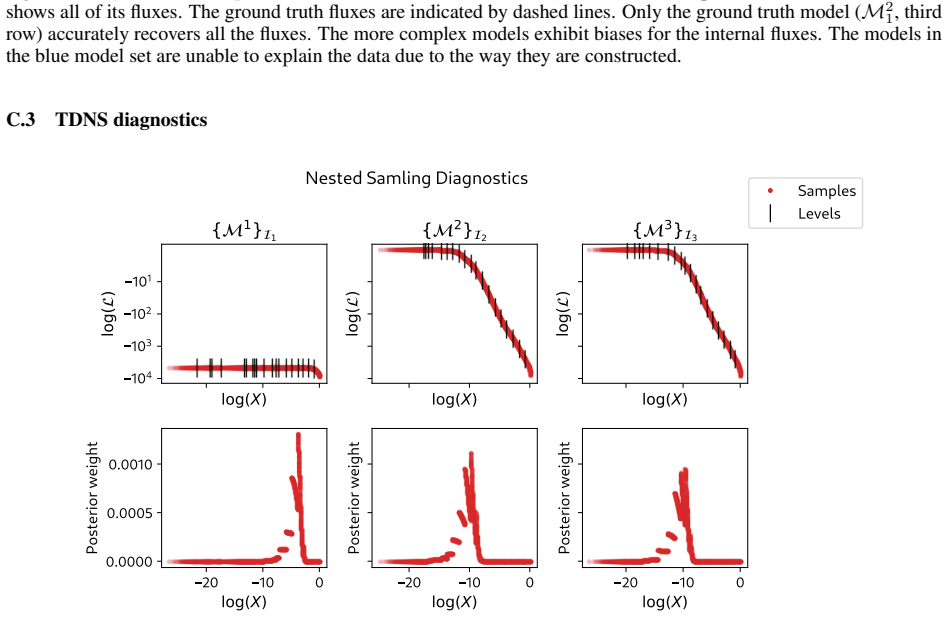
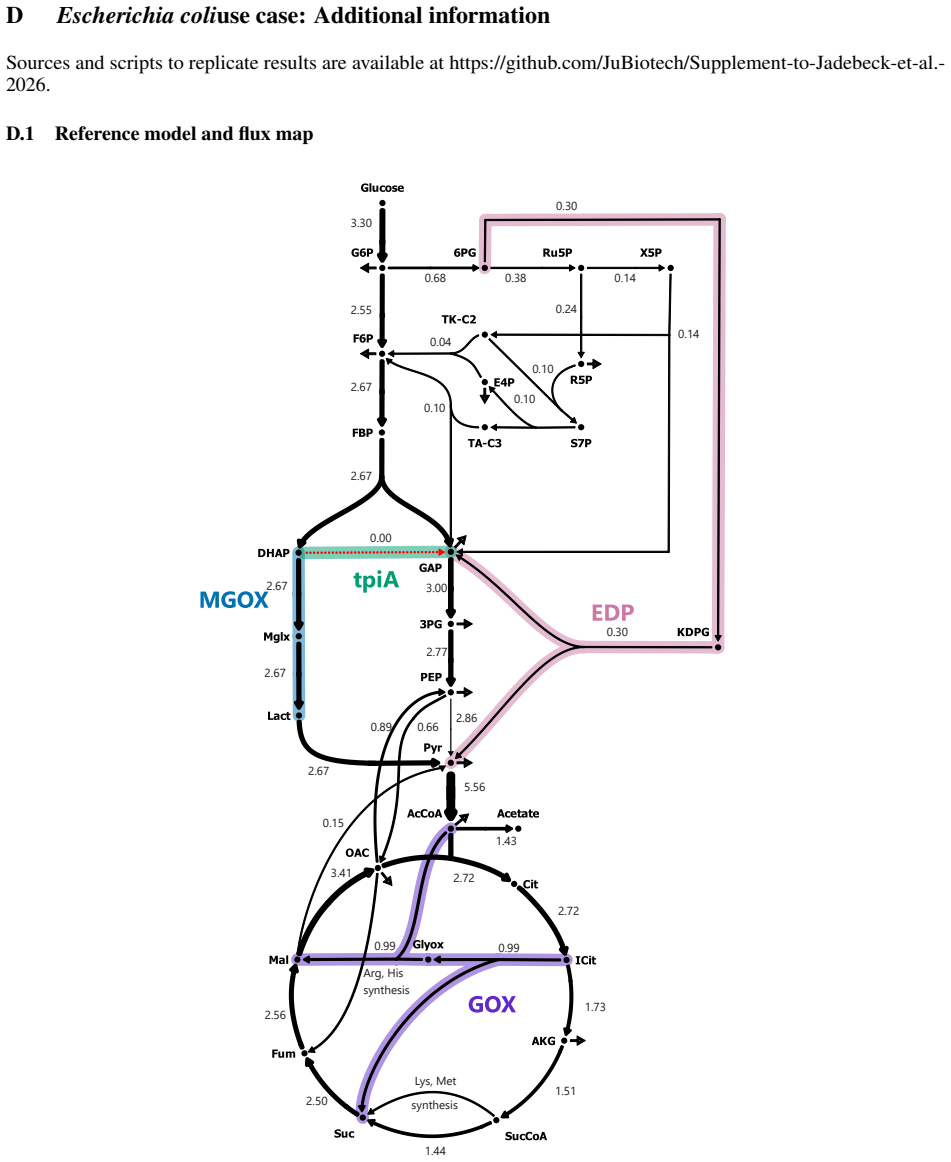
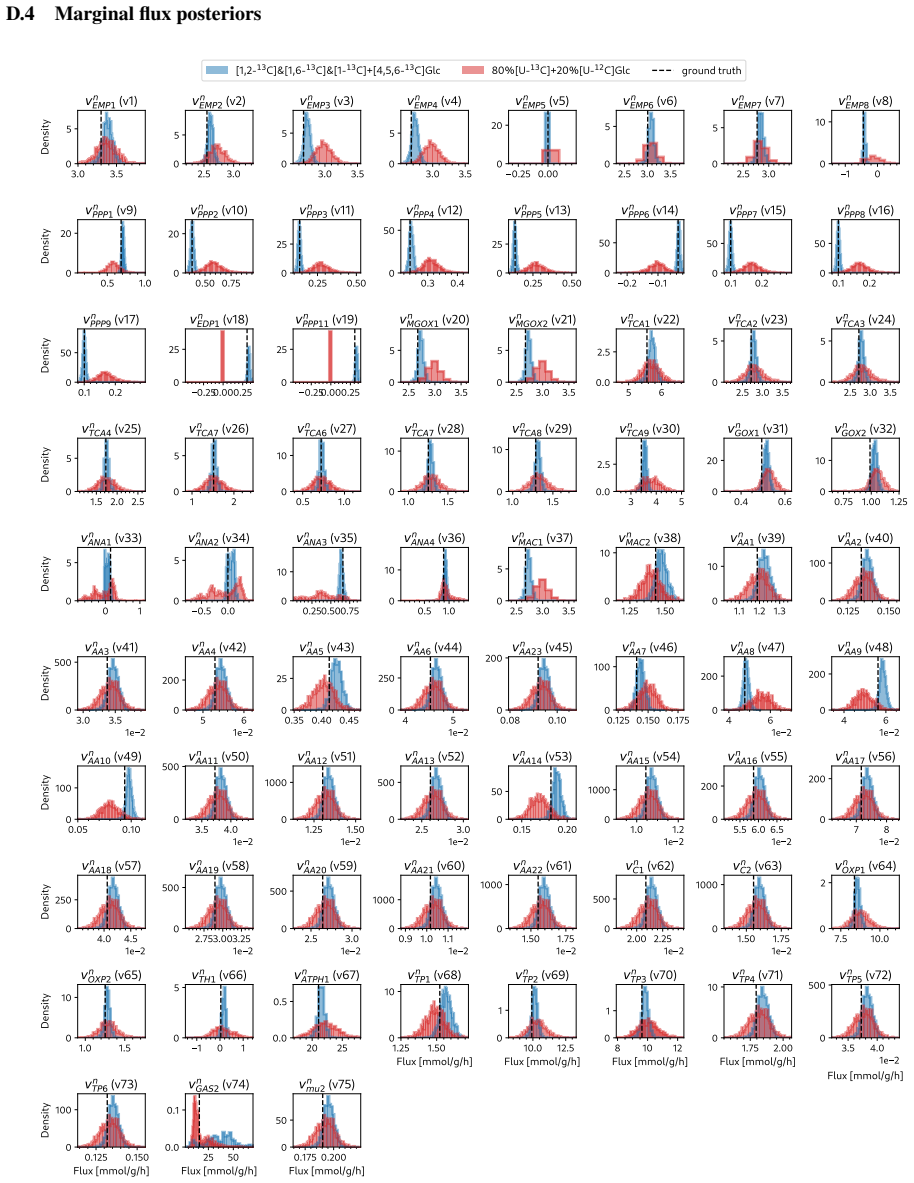
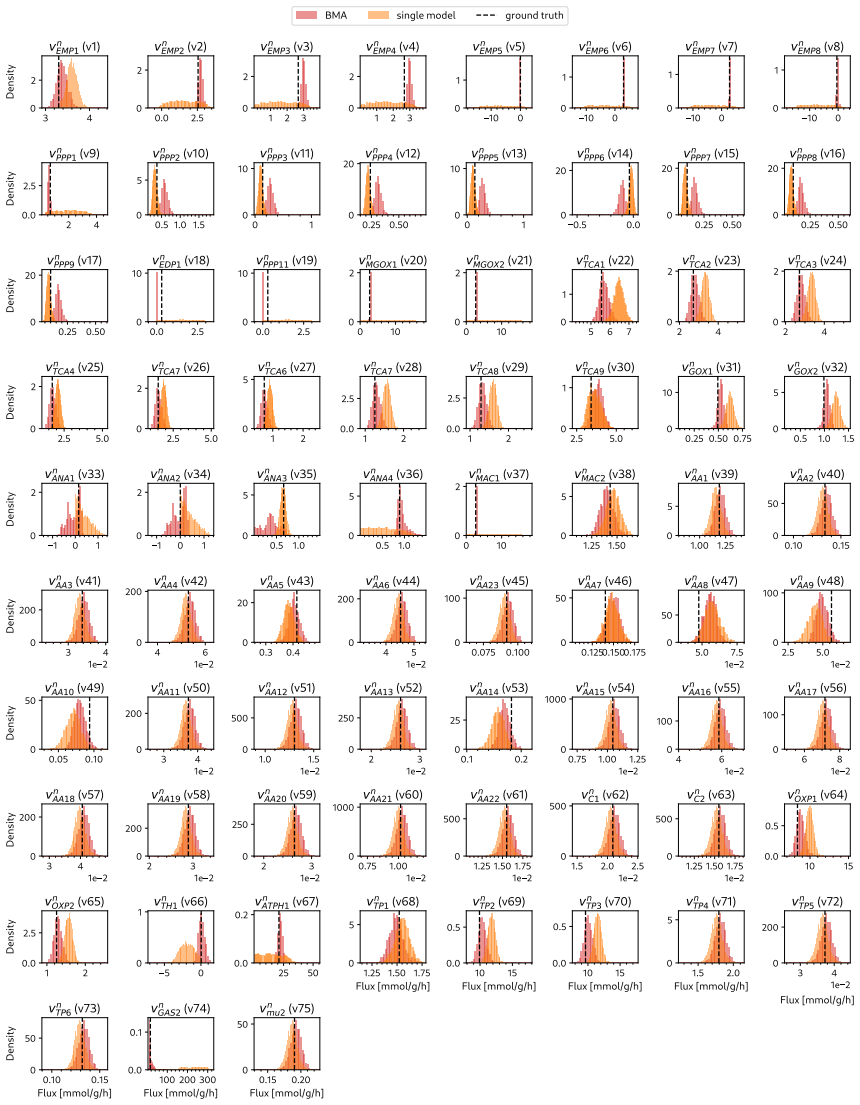




read the original abstract
Accurate quantification of intracellular metabolic fluxes is central to systems biology and biotechnology. Flux estimation relies on biochemical network models, with $^{13}$C metabolic flux analysis (MFA) being the state-of-the-art approach. However, isotope labeling data are often insufficient to uniquely support a single network formulation. In such cases, flux estimates become model-dependent, highlighting the need for methods that explicitly account for structural uncertainty. Bayesian model averaging (BMA) provides a principled framework for this purpose, but its application to $^{13}$C-MFA has so far been restricted to uncertainty in reaction bidirectionality within fixed network topologies. We introduce a scalable Bayesian inference framework for $^{13}$C-MFA, Bayesian model set averaging, that applies BMA to encompass uncertainty in reactions and pathways. Our approach combines reversible jump Markov chain Monte Carlo for trans-dimensional exploration of model spaces with diffusive nested sampling for robust estimation of model evidences, enabling averaging over large families of metabolic network models. Using illustrative and application-scale synthetic case studies, we demonstrate that the method yields robust flux estimates, reveals when multiple network configurations are statistically indistinguishable, and recovers data-supported model structures. Importantly, rather than committing to a single model, the framework manages structural uncertainty: under limited data, competing models are retained, whereas increasing data informativeness improved model and flux recovery. The approach scales to billions of model variants, providing a practical foundation for uncertainty- and misspecification-aware quantitative flux inference in $^{13}$C-MFA.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a Bayesian model set averaging framework for 13C metabolic flux analysis. It combines reversible jump MCMC for trans-dimensional exploration of network model spaces with diffusive nested sampling for model evidence estimation, enabling averaging over structural uncertainties (reactions and pathways) rather than fixing a single topology. The central claims are that the method yields robust flux estimates, identifies statistically indistinguishable models, recovers data-supported structures on synthetic cases, and scales practically to model spaces containing billions of variants.
Significance. If the sampling methods are shown to mix and converge reliably, the framework would address a recognized limitation in 13C-MFA by providing a principled way to propagate structural model uncertainty into flux estimates, which is particularly relevant when labeling data are limited.
major comments (1)
- [Abstract] Abstract: the central claim that the approach 'scales to billions of model variants' and enables practical averaging over large families rests on the unvalidated assumption that RJMCMC mixes sufficiently and that diffusive nested sampling returns reliable marginal likelihoods in combinatorially enormous spaces. No acceptance rates, effective sample sizes, convergence diagnostics, or timing results are reported for the application-scale synthetic cases, making it impossible to confirm that the reported flux recovery and model selection are not artifacts of poor exploration or biased evidence estimates.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback on our manuscript. The single major comment raises an important point about the need for explicit sampling diagnostics to support the scalability claims. We address this below and will incorporate the requested information in a revised version.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that the approach 'scales to billions of model variants' and enables practical averaging over large families rests on the unvalidated assumption that RJMCMC mixes sufficiently and that diffusive nested sampling returns reliable marginal likelihoods in combinatorially enormous spaces. No acceptance rates, effective sample sizes, convergence diagnostics, or timing results are reported for the application-scale synthetic cases, making it impossible to confirm that the reported flux recovery and model selection are not artifacts of poor exploration or biased evidence estimates.
Authors: We agree that convergence diagnostics are essential to substantiate the scalability claims. The current manuscript does not report acceptance rates, effective sample sizes, Gelman-Rubin statistics, or timing results for the application-scale synthetic cases. We will add these metrics in a revised Methods section and/or supplementary material, including trace plots, autocorrelation times, and evidence of adequate mixing for both RJMCMC and diffusive nested sampling on the largest model spaces examined. This addition will directly address the concern and allow readers to evaluate the reliability of the reported flux and model recovery results. revision: yes
Circularity Check
No circularity; framework combines independent sampling methods
full rationale
The paper presents a methodological combination of reversible jump MCMC for trans-dimensional model exploration and diffusive nested sampling for evidence estimation. No derivation step reduces by construction to a fitted parameter, self-citation load-bearing premise, or renamed input. Synthetic case studies are described as independent validation. The central claim of scalability is presented as an empirical demonstration rather than a tautological re-expression of inputs. No self-definitional, fitted-input, or uniqueness-imported patterns appear in the provided abstract or described structure.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
M. R. Antoniewicz. A guide to 13 C metabolic flux analysis for the cancer biologist . Experimental & molecular medicine, 50 0 (4): 0 19, 2018. doi:10.1038/s12276-018-0060-y
-
[2]
G. Ashton, N. Bernstein, J. Buchner, X. Chen, G. Cs \' a nyi, A. Fowlie, F. Feroz, M. Griffiths, W. Handley, M. Habeck, E. Higson, M. Hobson, A. Lasenby, D. Parkinson, L. B. P \' a rtay, M. Pitkin, D. Schneider, J. S. Speagle, L. South, J. Veitch, P. Wacker, D. J. Wales, and D. Yallup. Nested sampling for physical scientists . Nature Reviews Methods Prime...
-
[3]
D. J. Beste, K. N \" o h, S. Niedenf \" u hr, T. A. Mendum, N. D. Hawkins, J. L. Ward, M. H. Beale, W. Wiechert, and J. McFadden. 13 C - F lux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chemistry & Biology, 20 0 (8): 0 1012--1021, 2013. doi:10.1016/j.chembiol.2013.06.012
-
[4]
K. Borah Slater , M. Bey , Y. Xu, J. Barber, C. Costa, J. Newcombe, A. Theorell, M. J. Bailey, D. J. V. Beste, J. McFadden, and K. N \" o h. One‐shot 13 C 15 N ‐metabolic flux analysis for simultaneous quantification of carbon and nitrogen flux. Molecular Systems Biology, 19 0 (3), 2023. doi:10.15252/msb.202211099
-
[6]
B. J. Brewer and D. Foreman-Mackey. DNest4 : Diffusive nested sampling in C++ and P ython. Journal of Statistical Software, 86 0 (7): 0 1--33, 2018. doi:10.18637/jss.v086.i07
-
[7]
B. J. Brewer, L. B. P \'a rtay, and G. Cs \'a nyi. Diffusive nested sampling. Statistics and Computing, 21 0 (4): 0 649--656, 2011
2011
-
[8]
B. J. Brewer, D. Huijser, and G. F. Lewis. Trans-dimensional Bayesian inference for gravitational lens substructures . Monthly Notices of the Royal Astronomical Society, 455 0 (2): 0 1819--1829, 2015. doi:10.1093/mnras/stv2370
-
[9]
S. S. Fong, A. Nanchen, B. O. Palsson, and U. Sauer. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. Journal of Biological Chemistry, 281: 0 8024--8033, 2006. doi:10.1074/jbc.M510016200
-
[10]
A. Fowlie, W. Handley, and L. Su. Nested sampling with plateaus. Monthly Notices of the Royal Astronomical Society, 503 0 (1): 0 1199--1205, 2021. doi:10.1093/mnras/stab590
-
[11]
Z. Gong, J. Chen, X. Jiao, H. Gong, D. Pan, L. Liu, Y. Zhang, and T. Tan. Genome-scale metabolic network models for industrial microorganisms metabolic engineering: Current advances and future prospects . Biotechnology Advances, 72: 0 108319, 2024. doi:10.1016/j.biotechadv.2024.108319
-
[12]
P. J. Green. Reversible Jump Markov Chain Monte Carlo computation and Bayesian model determination . Biometrika, 82 0 (4): 0 711--732, 1995
1995
-
[13]
J. A. Hoeting, D. Madigan, A. E. Raftery, and C. T. Volinsky. Bayesian model averaging: a tutorial . Statistical Science, 14 0 (4): 0 382 -- 417, 1999. doi:10.1214/ss/1009212519
-
[14]
Z. Hu, A. Baryshnikov, and W. Handley. AEONS: approximating the end of nested sampling . Monthly Notices of the Royal Astronomical Society, 532 0 (4): 0 4035--4049, 2024. doi:10.1093/mnras/stae1754
-
[15]
J. F. Jadebeck, A. Theorell, S. Leweke, and K. Nöh. HOPS : high-performance library for (non-)uniform sampling of convex-constrained models. Bioinformatics, 37 0 (12): 0 1776--1777, 2020. doi:10.1093/bioinformatics/btaa872
-
[16]
J. F. Jadebeck, W. Wiechert, and K. N \"o h. Practical sampling of constraint-based models: Optimized thinning boosts CHRR performance. PLOS Computational Biology, 19 0 (8): 0 e1011378, 2023
2023
-
[17]
J. F. Jadebeck, W. Wiechert, and K. Nöh. Trans-dimensional diffusive nested sampling for metabolic network inference. Physical Sciences Forum, 12 0 (1): 0 5, 2025. doi:10.3390/psf2025012005
-
[18]
J. Kappelmann, W. Wiechert, and S. Noack. Cutting the Gordian Knot: Identifiability of anaplerotic reactions in Corynebacterium glutamicum by means of 13 C-metabolic flux analysis . Biotechnology and Bioengineering, 113 0 (3): 0 661--674, 2016. doi:10.1002/bit.25833
-
[19]
R. E. Kass and A. E. Raftery. Bayes factors. Journal of the American Statistical Association, 90 0 (430): 0 773--795, 1995. doi:10.1080/01621459.1995.10476572
-
[20]
R. W. Leighty and M. R. Antoniewicz. COMPLETE-MFA: Complementary parallel labeling experiments technique for metabolic flux analysis . Metabolic Engineering, 20: 0 49--55, 2013. doi:10.1016/j.ymben.2013.08.006
-
[21]
N. Linden-Santangeli and P. Rangamani. Increasing certainty in systems biology models using Bayesian multimodel inference . Nature Communications, 16: 0 7416, 2025. doi:10.1038/s41467-025-62415-4
-
[22]
L. Liu, D. Ding, H. Wang, X. Ren, S. Y. Lee, and D. Zhang. Balancing cell growth and product synthesis for efficient microbial cell factories . Advanced Science, 12 0 (40), 2025. doi:10.1002/advs.202510649
-
[23]
C. P. Long and M. R. Antoniewicz. High-resolution 13 C metabolic flux analysis . Nature Protocols, 14 0 (10): 0 2856--2877, 2019. doi:10.1038/s41596-019-0204-0
-
[24]
D. J. MacKay. Information Theory, Inference, and Learning Algorithms . Cambridge University Press, Cambridge, 2008. ISBN 0521642981. doi:10.2277/0521642981
-
[25]
J. McFadden. Razor sharp: The role of Occam's razor in science . Annals of the New York Academy of Sciences, 1530 0 (1): 0 8--17, 2023. doi:https://doi.org/10.1111/nyas.15086
-
[26]
S. Niedenf \" u hr, W. Wiechert, and K. N \" o h. How to measure metabolic fluxes: A taxonomic guide for 13 C fluxomics . Current Opinion in Biotechnology, 34: 0 82--90, 2015. doi:10.1016/j.copbio.2014.12.003
-
[27]
T. Nishikawa, N. Gulbahce, and A. E. Motter. Spontaneous reaction silencing in metabolic optimization . PLoS computational biology, 4 0 (12): 0 e1000236, 2008. doi:10.1371/journal.pcbi.1000236
-
[28]
R. D. Paul, J. F. Jadebeck, A. Stratmann, W. Wiechert, and K. Nöh. hopsy - a methods marketplace for convex polytope sampling in python. Bioinformatics, 40 0 (7): 0 btae430, 2024. doi:10.1093/bioinformatics/btae430
-
[29]
U. Sauer. Metabolic networks in motion: 13 C -based flux analysis. Molecular Systems Biology, 2: 0 62, 2006. doi:10.1038/msb4100109
-
[30]
D. Schittenhelm and P. Wacker. Nested sampling and likelihood plateaus. arXiv preprint arXiv:2005.08602, 2021
-
[31]
J. Skilling. Nested sampling for general Bayesian computation . Bayesian Analysis, 1 0 (4): 0 833 -- 859, 2006. doi:10.1214/06-BA127
-
[32]
A. Stratmann, M. Bey , J. F. Jadebeck, W. Wiechert, and K. N \" o h. 13CFLUX - third-generation high-performance engine for isotopically (non)stationary 13C metabolic flux analysis . Bioinformatics, 41 0 (12): 0 6, 2025. doi:10.1093/bioinformatics/btaf630
-
[33]
N. Sundqvist, N. Grankvist, J. Watrous, J. Mohit, R. Nilsson, and G. Cedersund. Validation-based model selection for 13 C metabolic flux analysis with uncertain measurement errors . PLOS Computational Biology, 18 0 (4): 0 e1009999, 2022. doi:10.1371/journal.pcbi.1009999
-
[34]
A. Theorell, S. Leweke, W. Wiechert, and K. Nöh. To be certain about the uncertainty: Bayesian statistics for 13 C metabolic flux analysis . Biotechnology and Bioengineering, 114 0 (11): 0 2668--2684, 2017. doi:https://doi.org/10.1002/bit.26379
-
[35]
A. Theorell, J. F. Jadebeck, K. N \" o h, and J. Stelling. PolyRound: polytope rounding for random sampling in metabolic networks . Bioinformatics, 38 0 (2): 0 566--567, 2022. doi:10.1093/bioinformatics/btab552
-
[36]
A. Theorell, J. F. Jadebeck, W. Wiechert, J. McFadden, and K. N \" o h. Rethinking 13 C-metabolic flux analysis – The Bayesian way of flux inference . Metabolic Engineering, 83: 0 137--149, 2024. doi:10.1016/j.ymben.2024.03.005
-
[37]
Wiechert
W. Wiechert. The thermodynamic meaning of metabolic exchange fluxes. Biophysical journal, 93 0 (6): 0 2255--2264, 2007
2007
-
[39]
W. Wiechert and K. N \" o h. Quantitative metabolic flux analysis based on isotope labeling . In J. Nielsen, G. Stephanopoulos, and S. Y. Lee, editors, Metabolic Engineering: Concepts and Applications, chapter 3, pages 73--136. Wiley, 2021. doi:10.1002/9783527823468.ch3
-
[40]
W. Wiechert, C. Siefke, A. A. de Graaf, and A. Marx. Bidirectional reaction steps in metabolic networks: II. Flux estimation and statistical analysis . Biotechnology and Bioengineering, 55 0 (1): 0 118–135, 1997. doi:10.1002/(sici)1097-0290(19970705)55:1<118::aid-bit13>3.0.co;2-i
-
[41]
N. Zamboni, S.-M. Fendt, M. R \" u hl, and U. Sauer. 13 C-based metabolic flux analysis . Nature Protocols, 4: 0 878--892, 2009. doi:10.1038/nprot.2009.58
-
[42]
Antoniewicz, M.R. (2018). A guide to 13 C metabolic flux analysis for the cancer biologist. Exp Mol Med , 50: 19
2018
-
[43]
Beste, D.J. et al. (2013). 13 C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem Biol , 20: 1012--1021
2013
-
[44]
Borah Slater, K. et al. (2023). One-shot ^ 13 C ^ 15 N metabolic flux analysis for simultaneous quantification of carbon and nitrogen flux. Mol Syst Biol, 19 0 (3): 0 e11099
2023
-
[45]
Brewer, B.J. (2015). Inference for trans-dimensional Bayesian models with diffusive nested sampling. arXiv preprint arXiv:1411.3921
work page internal anchor Pith review Pith/arXiv arXiv 2015
-
[46]
and Foreman-Mackey, D
Brewer, B.J. and Foreman-Mackey, D. (2018). DNest4 : diffusive nested sampling in C++ and Python. J Stat Softw , 86: 1--33
2018
-
[47]
Brewer, B.J. et al. (2015). Trans-dimensional Bayesian inference for gravitational lens substructures. Mon Not R Astron Soc , 455: 1819--1829
2015
-
[48]
Brewer, B.J. et al. (2011). Diffusive nested sampling. Stat Comput , 21: 649--656
2011
-
[49]
Fong, S.S. et al. (2006). Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem , 281: 8024--8033
2006
-
[50]
Gong, Z. et al. (2024). Genome-scale metabolic network models for industrial microorganisms metabolic engineering: current advances and future prospects. Biotechnol Adv , 72: 108319
2024
-
[51]
Green, P.J. (1995). Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika , 82: 711--732
1995
-
[52]
Hoeting, J.A. et al. (1999). Bayesian model averaging: a tutorial. Stat Sci , 14: 382--417
1999
-
[53]
Jadebeck, J.F. et al. (2020). HOPS : high-performance library for (non-)uniform sampling of convex-constrained models. Bioinformatics , 37: 1776--1777
2020
-
[54]
Jadebeck, J.F. et al. (2023). Practical sampling of constraint-based models: optimized thinning boosts CHRR performance. PLoS Comput Biol , 19: e1011378
2023
-
[55]
Kappelmann, J. et al. (2016). Cutting the Gordian knot: identifiability of anaplerotic reactions in Corynebacterium glutamicum by means of 13 C-metabolic flux analysis. Biotechnol Bioeng , 113: 661--674
2016
-
[56]
and Raftery, A.E
Kass, R.E. and Raftery, A.E. (1995). Bayes factors. J Am Stat Assoc , 90: 773--795
1995
-
[57]
and Antoniewicz, M.R
Leighty, R.W. and Antoniewicz, M.R. (2013). COMPLETE-MFA : complementary parallel labeling experiments technique for metabolic flux analysis. Metab Eng , 20: 49--55
2013
-
[58]
and Rangamani, P
Linden-Santangeli, N. and Rangamani, P. (2025). Increasing certainty in systems biology models using Bayesian multimodel inference . Nat Com , 16: 7416
2025
-
[59]
Liu, L. et al. (2025). Balancing cell growth and product synthesis for efficient microbial cell factories. Adv Sci , 12: e10649
2025
-
[60]
and Antoniewicz, M.R
Long, C.P. and Antoniewicz, M.R. (2019). High-resolution 13 C metabolic flux analysis. Nat Protoc , 14: 2856--2877
2019
-
[61]
MacKay, D.J.C. (2008). Information Theory, Inference, and Learning Algorithms . Cambridge University Press, Cambridge
2008
-
[62]
McFadden, J. (2023). Razor sharp: the role of Occam's razor in science. Ann N Y Acad Sci , 1530: 8--17
2023
-
[63]
Niedenf \"u hr, S. et al. (2015). How to measure metabolic fluxes: a taxonomic guide for 13 C fluxomics. Curr Opin Biotechnol , 34: 82--90
2015
-
[64]
Nishikawa, T. et al. (2008). Spontaneous reaction silencing in metabolic optimization. PLoS Comput Biol , 4: e1000236
2008
-
[65]
Paul, R.D. et al. (2024). hopsy : a methods marketplace for convex polytope sampling in Python. Bioinformatics , 40: btae430
2024
-
[66]
Sauer, U. (2006). Metabolic networks in motion: 13 C-based flux analysis. Mol Syst Biol , 2: 62
2006
- [67]
-
[68]
Sundqvist, N. et al. (2022). Validation-based model selection for 13 C metabolic flux analysis with uncertain measurement errors. PLoS Comput Biol , 18: e1009999
2022
-
[69]
Theorell, A. et al. (2022). PolyRound : polytope rounding for random sampling in metabolic networks. Bioinformatics , 38: 566--567
2022
-
[70]
Theorell, A. et al. (2024). Rethinking 13 C metabolic flux analysis -- the Bayesian way of flux inference. Metab Eng , 83: 137--149
2024
-
[71]
Theorell, A. et al. (2017). To be certain about the uncertainty: Bayesian statistics for 13 C metabolic flux analysis. Biotechnol Bioeng , 114: 2668--2684
2017
-
[72]
and N \"o h, K
Theorell, A. and N \"o h, K. (2020). Reversible jump MCMC for multi-model inference in metabolic flux analysis. Bioinformatics , 36: 232--240
2020
-
[73]
Wiechert, W. (2007). The thermodynamic meaning of metabolic exchange fluxes. Biophys J , 93: 2255--2264
2007
-
[74]
and de Graaf, A.A
Wiechert, W. and de Graaf, A.A. (1997). Bidirectional reaction steps in metabolic networks. Part I. Modeling and simulation of carbon isotope labeling experiments. Biotechnol Bioeng , 55: 101--117
1997
-
[75]
and N \"o h, K
Wiechert, W. and N \"o h, K. (2021). Quantitative metabolic flux analysis based on isotope labeling. In: Nielsen, J., Stephanopoulos, G. and Lee, S.-Y. (eds), Metabolic Engineering: Concepts and Applications . Wiley-VCH, Weinheim, Germany, pp.\ 73--136
2021
-
[76]
Wiechert, W. et al. (1997). Bidirectional reaction steps in metabolic networks: II. Flux estimation and statistical analysis. Biotechnol Bioeng , 55: 118--135
1997
-
[77]
Zamboni, N. et al. (2009). 13 C-based metabolic flux analysis. Nat Protoc , 4: 878--892
2009
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.