OCOO-T : A Simple and Scalable Virtual Cell Model for Transcriptional Perturbation Response Prediction
Pith reviewed 2026-06-27 05:20 UTC · model grok-4.3
The pith
A vanilla Transformer with flow-matching and adaptive normalization predicts single-cell transcriptional responses to perturbations at state-of-the-art accuracy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
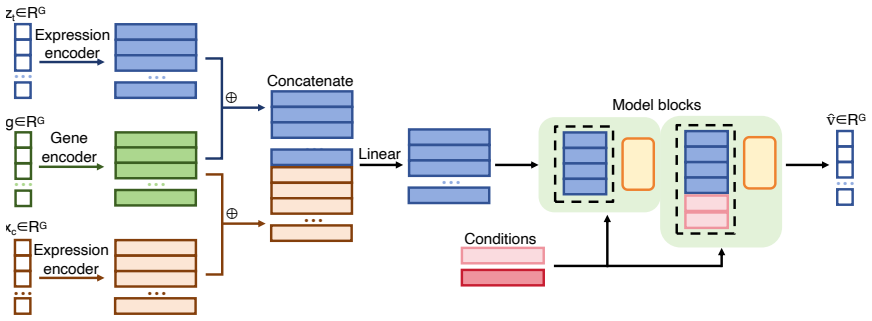
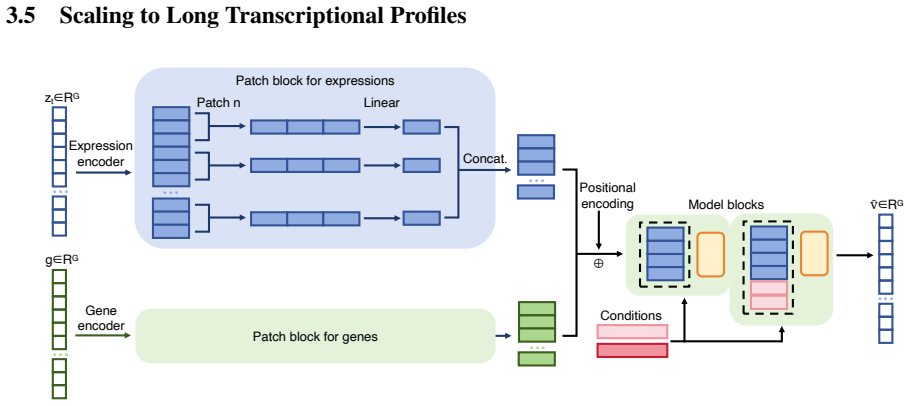
OCOO-T formulates transcriptional perturbation response prediction as a continuous-time flow-matching denoising task performed by a vanilla Transformer that operates directly on continuous gene-expression vectors; perturbation embeddings, dosage, and cell specificity are supplied solely through adaptive layer normalization and in-context tokens, enabling state-of-the-art accuracy across diverse perturbations and cell types on Tahoe100M, Replogle, and PBMC benchmarks together with linear scaling to long profiles through patching and depatching.
What carries the argument
Vanilla Transformer stack performing flow-matching denoising on continuous gene-expression profiles, conditioned by adaptive layer normalization and in-context tokens.
If this is right
- The model scales linearly to full-length transcriptional profiles by patching and depatching cellular contexts.
- Performance remains competitive across genetic, chemical, and cytokine perturbations as well as multiple cell types.
- Architectural complexity can be reduced while preserving or improving accuracy on existing single-cell perturbation benchmarks.
- In-silico cellular simulation becomes feasible at larger scale because the design avoids dedicated encoder-decoder modules.
Where Pith is reading between the lines
- If the minimalist conditioning proves sufficient, explicit gene-interaction graphs may be unnecessary for many perturbation-prediction tasks.
- The same patching strategy could be tested on other high-dimensional single-cell modalities such as chromatin accessibility or protein abundance.
- Training cost and iteration speed for virtual-cell models would drop if the vanilla-Transformer baseline continues to match specialized architectures.
Load-bearing premise
That perturbation type, dosage, and cell identity supplied only through adaptive layer normalization and in-context tokens are sufficient to capture relevant biological response dynamics without gene-interaction priors or hierarchical encoders.
What would settle it
A new benchmark dataset containing strong, previously unseen gene-regulatory interactions where any method that explicitly encodes those interactions significantly outperforms OCOO-T on held-out perturbations.
Figures




read the original abstract
Predicting single-cell transcriptional responses to genetic, chemical and cytokine perturbations is a fundamental challenge in computational biology and AI Virtual Cell (AIVC) modeling, with direct implications for drug discovery and the elucidation of gene regulatory networks. Existing approaches often rely on auxiliary cell-state encoders, hierarchical variational autoencoders, dedicated Transformer encoder-decoder modules, or gene-interaction priors to compress high-dimensional expression profiles into latent representations. While effective, these designs increase architectural complexity and may limit scalability and generalizability. This paper introduces OCOO-T, a minimalist flow-matching-based AIVC model for transcriptional perturbation response prediction. OCOO-T utilizes a vanilla Transformer stack that operates directly on continuous gene expression profiles and formulates perturbation response prediction as a continuous-time denoising process. Perturbation embeddings, dosage information, and cell-line/cell-type specificity are integrated through adaptive layer normalization and in-context tokens. Comprehensive evaluations on Tahoe100M, Replogle, and PBMC benchmarks demonstrate that OCOO-T achieves state-of-the-art performance across diverse perturbations and cell types while effectively scaling to long transcriptional profiles through patching and depatching of cellular contexts. By leveraging the simplicity of Transformer-based denoising for single-cell omics, OCOO-T provides an effective and scalable framework for in-silico cellular simulation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces OCOO-T, a minimalist flow-matching-based virtual cell model that uses a vanilla Transformer operating directly on continuous gene expression profiles to predict single-cell transcriptional responses to genetic, chemical, and cytokine perturbations. Perturbation embeddings, dosage, and cell specificity are incorporated via adaptive layer normalization and in-context tokens, with patching/depatching for scalability to long profiles. The central claim is that this simple architecture achieves state-of-the-art performance on the Tahoe100M, Replogle, and PBMC benchmarks across diverse perturbations and cell types.
Significance. If the performance claims hold with proper validation, this would be significant for AIVC modeling by showing that standard flow-matching and Transformer components can suffice without auxiliary encoders, hierarchical VAEs, or gene-interaction priors, potentially improving scalability and reproducibility. The emphasis on a parameter-light design using established techniques is a strength for the field.
major comments (1)
- [Abstract] Abstract: the assertion of state-of-the-art performance on Tahoe100M, Replogle, and PBMC benchmarks provides no quantitative metrics, baseline details, error analysis, or statistical comparisons, which is load-bearing for the central empirical claim and prevents verification of the reported improvements.
Simulated Author's Rebuttal
We thank the referee for their review and constructive comment. We address the concern about the abstract below and will incorporate the suggested changes in the revised manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the assertion of state-of-the-art performance on Tahoe100M, Replogle, and PBMC benchmarks provides no quantitative metrics, baseline details, error analysis, or statistical comparisons, which is load-bearing for the central empirical claim and prevents verification of the reported improvements.
Authors: We agree that the abstract would be strengthened by including key quantitative metrics to support the SOTA claim. In the revised version, we will add concise performance highlights (e.g., primary metrics and baseline comparisons on each benchmark) drawn directly from the results tables, while preserving the abstract's length and readability. This addresses the verification concern without altering the manuscript's core claims. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper introduces OCOO-T as a minimalist flow-matching Transformer model for perturbation response prediction, relying on standard components (vanilla Transformer, adaptive layer norm, in-context tokens, patching) and reports empirical SOTA results on external benchmarks (Tahoe100M, Replogle, PBMC). No equations, derivations, or predictions are presented that reduce by construction to fitted parameters or self-defined quantities. No load-bearing self-citations, uniqueness theorems, or ansatzes imported from prior author work are invoked; the central claims rest on benchmark performance rather than internal definitional closure. This is a standard empirical modeling paper with no circular steps.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URLhttps://doi.org/10.1038/s41592-024-02201-0
Haotian Cui et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI.Nature Methods, 21(8):1470–1480, 2024. doi:10.1038/s41592-024-02201-0
-
[2]
Anish K. Adduri et al. Predicting cellular responses to perturbation across diverse contexts with State. bioRxiv, 2025. doi:10.1101/2025.06.26.661135. 17
-
[3]
Denoising Diffusion Probabilistic Models
Jonathan Ho, Ajay Jain, and Pieter Abbeel. Denoising Diffusion Probabilistic Models. InAdvances in Neural Information Processing Systems (NeurIPS), 2020
2020
-
[4]
Yaron Lipman, Ricky T. Q. Chen, Heli Ben-Hamu, Maximilian Nickel, and Matt Le. Flow Matching for Generative Modeling. InInternational Conference on Learning Representations (ICLR), 2023
2023
-
[5]
Sheng He et al. Squidiff: predicting cellular development and responses to perturbations using a diffusion model.Nature Methods, 2025. doi:10.1038/s41592-025-02877-y
-
[6]
Dominik Klein et al. CellFlow Enables Generative Single-Cell Phenotype Modeling with Flow Matching. bioRxiv, 2025. doi:10.1101/2025.04.11.648220
-
[7]
Zhaokang Liang, Shuyang Zhuang, Xiaoran Jiao, Weian Mao, Hao Chen, and Chunhua Shen. scPPDM: A Diffusion Model for Single-Cell Drug-Response Prediction.arXiv preprint arXiv:2510.11726, 2025
-
[8]
Chenglei Yu, Chuanrui Wang, Bangyan Liao, and Tailin Wu. scDFM: Distributional Flow Matching Model for Robust Single-Cell Perturbation Prediction.arXiv preprint arXiv:2602.07103, 2026
-
[9]
Xinyu Yuan, Xixian Liu, Ya Shi Zhang, Zuobai Zhang, Hongyu Guo, and Jian Tang. PerturbDiff: Functional Diffusion for Single-Cell Perturbation Modeling.arXiv preprint arXiv:2602.19685, 2026
-
[10]
Reddi, Aaditya Ramdas, Barnab ´as P ´oczos, Aarti Singh, and Larry Wasserman
Sashank J. Reddi, Aaditya Ramdas, Barnab ´as P ´oczos, Aarti Singh, and Larry Wasserman. On the Decreasing Power of Kernel and Distance Based Nonparametric Hypothesis Tests in High Dimensions. InProceedings of the Twenty-Ninth AAAI Conference on Artificial Intelligence (AAAI), 2015
2015
-
[11]
10 Million Human PBMCs in a Single Experiment
Parse Biosciences. 10 Million Human PBMCs in a Single Experiment. Dataset resource, 2023
2023
-
[12]
Jesse Zhang et al. Tahoe-100M: A Giga-Scale Single-Cell Perturbation Atlas for Context-Dependent Gene Function and Cellular Modeling.bioRxiv, 2025. doi:10.1101/2025.02.20.639398
-
[13]
Ajay Nadig, Joseph M. Replogle, Alexander N. Pogson, et al. Transcriptome-wide analysis of differential expression in perturbation atlases.Nature Genetics, 2025. doi:10.1038/s41588-025-02169-3
-
[14]
Mohammad Lotfollahi, F. Alexander Wolf, and Fabian J. Theis. scGen predicts single-cell perturbation responses.Nature Methods, 16(8):715–721, 2019. doi:10.1038/s41592-019-0494-8
-
[15]
Mohammad Lotfollahi et al. Predicting cellular responses to complex perturbations in high-throughput screens.Molecular Systems Biology, 19(6):e11517, 2023. doi:10.15252/msb.202211517
-
[16]
Leon Hetzel, Simon B¨ohm, Niki Kilbertus, Stephan G¨unnemann, Mohammad Lotfollahi, and Fabian J. Theis. Predicting cellular responses to novel drug perturbations at a single-cell resolution. InAdvances in Neural Information Processing Systems (NeurIPS), 2022
2022
-
[17]
Roohani, Kexin Huang, and Jure Leskovec
Yusuf H. Roohani, Kexin Huang, and Jure Leskovec. Predicting transcriptional outcomes of novel multigene perturbations with GEARS.Nature Biotechnology, 42:927–935, 2024. doi:10.1038/s41587- 023-01905-6
-
[18]
Large-scale foundation model on single-cell transcriptomics.Nature Methods, 21(8):1481–1491, 2024
Minsheng Hao et al. Large-scale foundation model on single-cell transcriptomics.Nature Methods, 21(8):1481–1491, 2024. doi:10.1038/s41592-024-02305-7
-
[19]
Ravindra, Lexi R
Chloe Wang, Mehran Karimzadeh, Neal G. Ravindra, Lexi R. Bounds, et al. X-Cell: Scaling Causal Perturbation Prediction Across Diverse Cellular Contexts via Diffusion Language Models.bioRxiv,
-
[20]
doi:10.64898/2026.03.18.712807. 18
-
[21]
Mudge et al
Jonathan M. Mudge et al. GENCODE 2025: reference gene annotation for human and mouse.Nucleic Acids Research, 53(D1):D966–D975, 2025
2025
-
[22]
Ding Bai et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics.Nature Communications, 17:2380, 2026. doi:10.1038/s41467-026-69102-y
-
[23]
Back to Basics: Let Denoising Generative Models Denoise
Tianhong Li and Kaiming He. Back to Basics: Let Denoising Generative Models Denoise.arXiv preprint arXiv:2511.13720, 2025
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[24]
Yusuf H. Roohani, Tony J. Hua, Po-Yuan Tung, Lexi R. Bounds, et al. Virtual Cell Challenge: Toward a Turing Test for the Virtual Cell.Cell, 188(13):3370–3374, 2025. doi:10.1016/j.cell.2025.06.008
-
[25]
Luke A. Gilbert et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154(2):442–451, 2013. doi:10.1016/j.cell.2013.06.044
-
[26]
Thomas M. Norman et al. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes.Science, 365(6455):786–793, 2019. doi:10.1126/science.aax4438
-
[27]
cell-eval: Comprehensive suite for evaluating perturbation prediction models
Arc Institute. cell-eval: Comprehensive suite for evaluating perturbation prediction models. GitHub repository, 2026
2026
-
[28]
Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines.Nature Methods, 22(8):1657–1661,
Constantin Ahlmann-Eltze, Wolfgang Huber, and Simon Anders. Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines.Nature Methods, 22(8):1657–1661,
-
[29]
doi:10.1038/s41592-025-02772-6
-
[30]
Weinstock, Alexander Battle, and Patrick Cahan
Eli Kernfeld, Yanyu Yang, Joshua S. Weinstock, Alexander Battle, and Patrick Cahan. A comparison of computational methods for expression forecasting.Genome Biology, 26:388, 2025. doi:10.1186/s13059- 025-03840-y. 19 Appendix A Training Details Backbone of the denoiser model is a 12-layer Transformer with hidden size 768 and 12 attention heads (head dimensi...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.