cuSBF: A Minimizer-Aware Bloom Filter for Genomic Sequence Data on Modern GPUs
Pith reviewed 2026-06-25 22:58 UTC · model grok-4.3
The pith
cuSBF implements a minimizer-aware Super Bloom Filter on GPUs that turns super-k-mer locality into scalable parallelism for genomic k-mer indexing.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
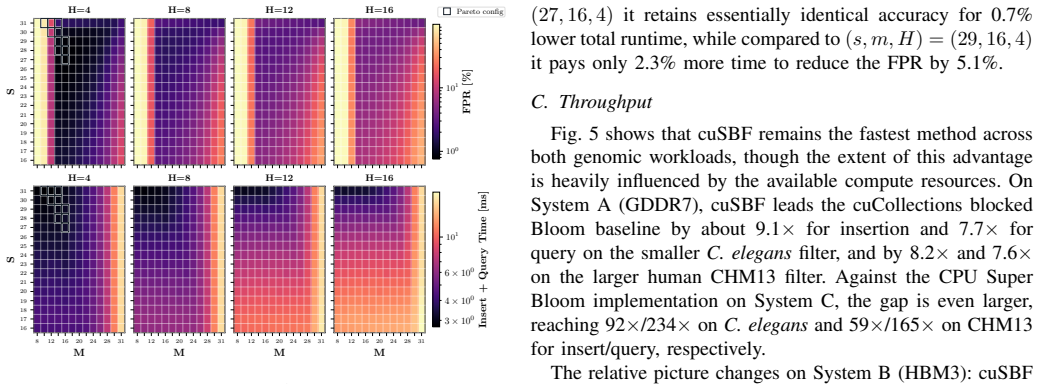
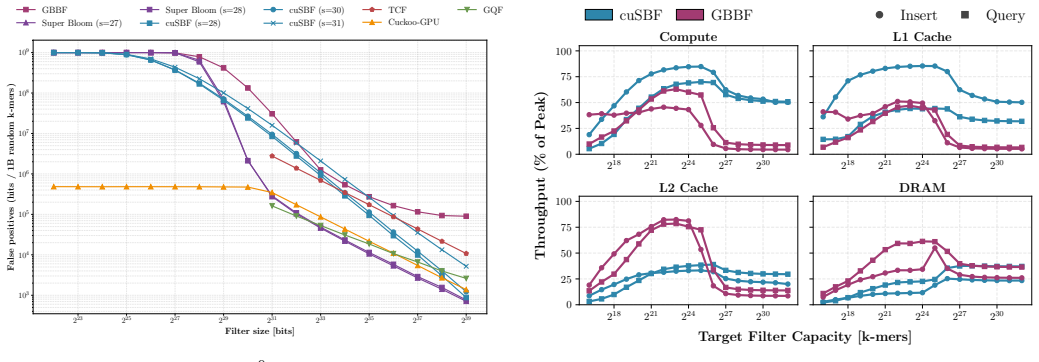
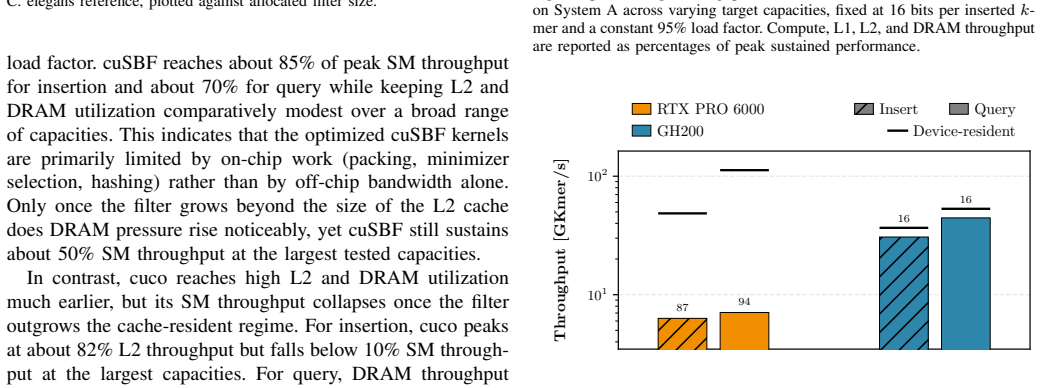
cuSBF merges sectorized shards, cooperative shared-memory tiling, warp-level shard sharing, and segmented warp reductions to convert super-k-mer locality into scalable GPU parallelism. On RTX PRO 6000 Blackwell hardware the design delivers up to 9.1x higher insertion throughput and 7.7x higher query throughput than the cuCollections blocked Bloom filter, up to 92x and 234x over a multi-threaded CPU Super Bloom reference, and 1.5-3400x over GPU dynamic structures such as Cuckoo and Quotient filters, while sustaining 85 percent streaming-multiprocessor utilization even for out-of-cache filters. A parameter sweep identifies (s=28, m=16, H=4) as Pareto-optimal for k=31 and produces lower false-p
What carries the argument
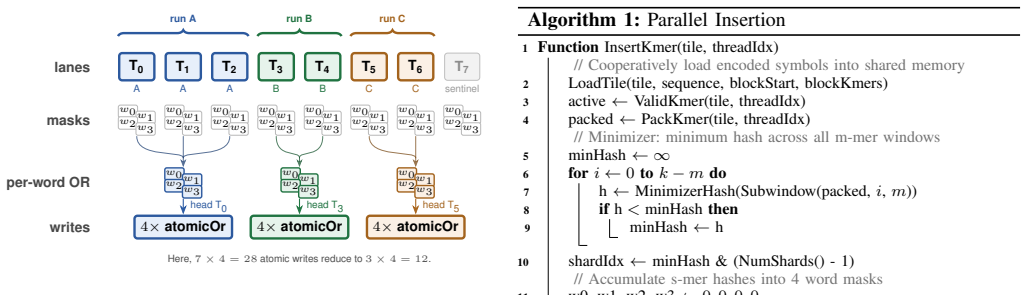
Sectorized shards combined with cooperative shared-memory tiling, warp-level shard sharing, and segmented warp reductions that exploit minimizer-guided super-k-mer locality.
If this is right
- The identified parameter set yields lower false-positive rates than cuCollections blocked Bloom filters at matched memory budgets.
- Sequence locality rather than raw memory bandwidth becomes the dominant performance factor for GPU-accelerated genomic indexing.
- The header-only library supports direct integration into existing sequence-analysis pipelines without external dependencies.
- Out-of-cache operation remains efficient, enabling indexing of datasets larger than GPU memory without proportional slowdown.
- The same architectural pattern outperforms multiple classes of dynamic approximate-membership-query structures by wide margins on genomic inputs.
Where Pith is reading between the lines
- The same tiling and reduction strategy could be tested on other locality-rich sequence tasks such as minimizer-based sketching or de Bruijn graph construction.
- Porting the design to newer GPU memory hierarchies or multi-GPU nodes would test whether the 85 percent utilization scales beyond single-device Blackwell and GH200 systems.
- Community extensions of the open-source code could adapt the sector size and hash parameters for non-human genomes with different repeat structures.
- The emphasis on warp-level reductions suggests analogous gains may exist for other irregular, locality-driven data structures in bioinformatics beyond Bloom filters.
Load-bearing premise
The locality properties of minimizer-guided super-k-mers in real genomic workloads can be reliably converted into scalable high-utilization GPU parallelism through sectorized shards, cooperative tiling, and warp reductions without hidden overheads that appear only at larger scale or different data distributions.
What would settle it
Measuring sustained streaming-multiprocessor utilization and throughput on genomic datasets whose minimizer distributions or super-k-mer lengths differ markedly from the evaluated workloads would show whether performance remains near 85 percent utilization or drops due to unaccounted overheads.
Figures



read the original abstract
Efficient genomic k-mer indexing depends on approximate membership query (AMQ) structures that must deliver high throughput, low false-positive rates (FPR), and modest memory footprints. The Super Bloom filter (SBF) is attractive for this scenario because minimizer-guided sharding and the Findere scheme exploit the redundancy of overlapping k-mers. However, those same features cause high per-k-mer compute cost, severe register pressure, and irregular memory accesses, which hinder an effective GPU implementation. We present cuSBF, an open-source, header-only CUDA library that implements SBF for sequence-native workloads. cuSBF's design merges sectorized shards, cooperative shared-memory tiling, warp-level shard sharing, and segmented warp reductions, turning super-k-mer locality into scalable GPU parallelism. Across real genomic workloads on RTX PRO 6000 Blackwell and GH200 systems, cuSBF achieves the highest throughput among all evaluated sequence-capable baselines. On the RTX PRO 6000, it surpasses the cuCollections blocked Bloom filter baseline by up to 9.1x for insertion and 7.7x for query, while reaching up to 92x and 234x speedups over the multi-threaded CPU Super Bloom reference implementation. It also outperforms GPU-based dynamic AMQs (Cuckoo, Two-Choice, Quotient filters) by 1.5-3400x depending on workload characteristics. A parameter sweep identifies (s = 28, m = 16, H = 4) as Pareto-optimal for k = 31, yielding significantly lower FPR than cuCollections at matched memory budgets. Crucially, cuSBF's architecture-aware design sustains 85% streaming multiprocessor utilization even for out-of-cache filters - proving that sequence locality, not raw bandwidth, is the key to GPU-accelerated genomic indexing.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents cuSBF, a header-only CUDA library implementing the Super Bloom Filter (SBF) for genomic k-mer indexing. It combines sectorized shards, cooperative shared-memory tiling, warp-level shard sharing, and segmented warp reductions to convert minimizer-guided super-k-mer locality into GPU parallelism. On RTX PRO 6000 and GH200, cuSBF reports the highest throughput among sequence-capable baselines, with speedups of up to 9.1x (insertion) and 7.7x (query) over cuCollections blocked Bloom filter, 92x/234x over multi-threaded CPU SBF, and 1.5-3400x over other GPU AMQs; a parameter sweep identifies (s=28, m=16, H=4) as Pareto-optimal for k=31 with lower FPR at matched memory, while sustaining 85% SM utilization even for out-of-cache filters.
Significance. If the throughput and utilization numbers are reproducible on the stated workloads and hardware, the work would supply a practical, open-source GPU primitive for high-throughput genomic AMQ that directly exploits sequence redundancy, potentially benefiting downstream k-mer indexing pipelines. The header-only design and explicit parameter identification are additional strengths.
major comments (2)
- [Abstract] Abstract: the central claim that sectorized shards + cooperative tiling + warp reductions convert super-k-mer locality into scalable 85% SM utilization (and the associated 9.1x/7.7x speedups) rests on an untested assumption that the locality pattern remains regular across larger or more diverse genomic inputs; no scaling experiment isolating shard imbalance or irregular access at scale is referenced, which is load-bearing for the generalization of the utilization result.
- [Abstract] Abstract: the statement that cuSBF 'proves that sequence locality, not raw bandwidth, is the key' is not supported by any described ablation that isolates bandwidth versus locality effects (e.g., synthetic random-access workloads versus genomic workloads at matched occupancy); this claim is load-bearing for the architectural takeaway.
minor comments (2)
- Abstract: workload sizes, number of distinct k-mers, and exact FPR values at the reported memory budgets are not stated; these should appear in the experimental section or a table for reproducibility.
- Abstract: the phrase 'real genomic workloads' is used without naming the datasets or providing a citation; add explicit references or dataset descriptions.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the abstract. We address each major comment below and indicate the revisions we will make to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that sectorized shards + cooperative tiling + warp reductions convert super-k-mer locality into scalable 85% SM utilization (and the associated 9.1x/7.7x speedups) rests on an untested assumption that the locality pattern remains regular across larger or more diverse genomic inputs; no scaling experiment isolating shard imbalance or irregular access at scale is referenced, which is load-bearing for the generalization of the utilization result.
Authors: Our reported results are based on multiple real genomic datasets of different sizes and characteristics, where we observed consistent 85% SM utilization. We acknowledge that these do not include dedicated scaling experiments that explicitly isolate shard imbalance or irregular access patterns at larger scales. To address this, we will add a new subsection with scaling experiments on larger inputs and synthetic workloads designed to stress shard balance in the revised manuscript. revision: yes
-
Referee: [Abstract] Abstract: the statement that cuSBF 'proves that sequence locality, not raw bandwidth, is the key' is not supported by any described ablation that isolates bandwidth versus locality effects (e.g., synthetic random-access workloads versus genomic workloads at matched occupancy); this claim is load-bearing for the architectural takeaway.
Authors: The statement is motivated by the observation that high utilization is maintained even when the filter exceeds on-chip cache capacity. However, we agree that the word 'proves' is not supported by a direct ablation isolating bandwidth from locality effects at matched occupancy. We will revise the abstract to remove or qualify this phrasing and add a short discussion clarifying the basis of the claim. revision: yes
Circularity Check
No circularity detected; claims rest on direct empirical benchmarks against external baselines
full rationale
The manuscript presents an engineering implementation (cuSBF) of an existing SBF structure with GPU-specific optimizations (sectorized shards, cooperative tiling, warp reductions) and reports measured throughput, FPR, and SM utilization from direct hardware benchmarks on RTX PRO 6000 and GH200 against named external baselines (cuCollections, CPU SBF, Cuckoo, etc.). No equations, fitted parameters, or predictions are claimed; performance numbers are not derived from internal quantities but measured. No self-citation chain is invoked to justify a uniqueness theorem or ansatz that would reduce the central result to prior author work. The design is presented as an architecture-aware engineering choice, not a mathematical necessity. This is the common case of a self-contained implementation paper whose results are externally falsifiable via re-execution.
Axiom & Free-Parameter Ledger
free parameters (1)
- s, m, H =
28,16,4
axioms (1)
- domain assumption Minimizer-guided sharding and Findere scheme exploit redundancy of overlapping k-mers
Reference graph
Works this paper leans on
-
[1]
Space/time trade-offs in hash coding with allowable errors,
B. H. Bloom, “Space/time trade-offs in hash coding with allowable errors,”Commun. ACM, vol. 13, no. 7, p. 422–426, Jul. 1970. [Online]. Available: https://doi.org/10.1145/362686.362692
-
[2]
F. Chang, J. Dean, S. Ghemawat, W. C. Hsieh, D. A. Wallach, M. Burrows, T. Chandra, A. Fikes, and R. E. Gruber, “Bigtable: A Distributed Storage System for Structured Data,”ACM Trans. Comput. Syst., vol. 26, no. 2, 2008. [Online]. Available: https: //doi.org/10.1145/1365815.1365816
-
[3]
Network Applications of Bloom Filters: A Survey,
A. Broder and M. Mitzenmacher, “Network Applications of Bloom Filters: A Survey,”Internet Mathematics, vol. 1, no. 4, pp. 485– 509, 2004. [Online]. Available: https://doi.org/10.1080/15427951.2004. 10129096
-
[4]
NGSReadsTreatment – A Cuckoo Filter-based Tool for Removing Duplicate Reads in NGS Data,
A. S. C. Gaia, P. H. C. G. de S ´a, M. S. de Oliveira, and A. A. d. O. Veras, “NGSReadsTreatment – A Cuckoo Filter-based Tool for Removing Duplicate Reads in NGS Data,”Scientific Reports, vol. 9, no. 1, p. 11681, Aug 2019. [Online]. Available: https://doi.org/10.1038/s41598-019-48242-w
-
[5]
Cache-, Hash- and Space- Efficient Bloom Filters,
F. Putze, P. Sanders, and J. Singler, “Cache-, Hash- and Space- Efficient Bloom Filters,” inExperimental Algorithms, C. Demetrescu, Ed. Springer, 2007, pp. 108–121
2007
-
[6]
cuCollections,
NVIDIA Corporation, “cuCollections,” Oct. 2026, gitHub repository, commit 8576506. [Online]. Available: https://github.com/NVIDIA/ cuCollections
2026
-
[7]
Optimizing Bloom Filters for Modern GPU Architectures,
D. J ¨unger, K. Kristensen, Y . Wang, X. Yu, and B. Schmidt, “Optimizing Bloom Filters for Modern GPU Architectures,” 2025. [Online]. Available: https://arxiv.org/abs/2512.15595
arXiv 2025
-
[8]
Super Bloom: Fast and precise filter for streaming k-mer queries,
E. Conchon-Kerjan, T. Rouz ´e, L. Robidou, F. Ingels, and A. Limasset, “Super Bloom: Fast and precise filter for streaming k-mer queries,” bioRxiv, 2026. [Online]. Available: https://www.biorxiv.org/content/ early/2026/03/19/2026.03.17.712354
2026
-
[9]
M. Roberts, W. Hayes, B. R. Hunt, S. M. Mount, and J. A. Yorke, “Reducing storage requirements for biological sequence comparison,” Bioinformatics, vol. 20, no. 18, pp. 3363–3369, 12 2004. [Online]. Available: https://doi.org/10.1093/bioinformatics/bth408
-
[10]
findere: Fast and Precise Approximate Membership Query,
L. Robidou and P. Peterlongo, “findere: Fast and Precise Approximate Membership Query,” inString Processing and Information Retrieval, T. Lecroq and H. Touzet, Eds. Cham: Springer International Publishing, 2021, pp. 151–163
2021
-
[11]
RapidGKC: GPU-Accelerated K- Mer Counting,
Y . Cheng, X. Sun, and Q. Luo, “RapidGKC: GPU-Accelerated K- Mer Counting,” in2024 IEEE 40th International Conference on Data Engineering (ICDE), May 2024, pp. 3810–3822
2024
-
[12]
Gerbil: a fast and memory-efficient k-mer counter with GPU-support,
M. Erbert, S. Rechner, and M. M ¨uller-Hannemann, “Gerbil: a fast and memory-efficient k-mer counter with GPU-support,”Algorithms for Molecular Biology, vol. 12, no. 1, p. 9, Mar 2017. [Online]. Available: https://doi.org/10.1186/s13015-017-0097-9
-
[13]
GPU Acceleration of Advanced k-mer Counting for Computational Genomics,
H. Li, A. Ramachandran, and D. Chen, “GPU Acceleration of Advanced k-mer Counting for Computational Genomics,” in2018 IEEE 29th International Conference on Application-specific Systems, Architectures and Processors (ASAP), July 2018, pp. 1–4
2018
-
[14]
High-Performance Filters for GPUs,
H. McCoy, S. Hofmeyr, K. Yelick, and P. Pandey, “High-Performance Filters for GPUs,” inProceedings of the 28th ACM SIGPLAN Annual Symposium on Principles and Practice of Parallel Programming, ser. PPoPP ’23. New York, NY , USA: ACM, 2023, p. 160–173. [Online]. Available: https://doi.org/10.1145/3572848.3577507
-
[15]
Quotient Filters: Approx- imate Membership Queries on the GPU,
A. Geil, M. Farach-Colton, and J. D. Owens, “Quotient Filters: Approx- imate Membership Queries on the GPU,” in2018 IEEE International Parallel and Distributed Processing Symposium (IPDPS), 2018, pp. 451– 462
2018
-
[16]
Cuckoo-GPU: Accelerating Cuckoo Filters on Modern GPUs,
T. Dortmann, M. Vieth, and B. Schmidt, “Cuckoo-GPU: Accelerating Cuckoo Filters on Modern GPUs,” 2026. [Online]. Available: https://arxiv.org/abs/2603.15486
arXiv 2026
-
[17]
Ultrafast search of all deposited bacterial and viral genomic data,
P. Bradley, H. C. den Bakker, E. P. C. Rocha, G. McVean, and Z. Iqbal, “Ultrafast search of all deposited bacterial and viral genomic data,” Nature Biotechnology, vol. 37, no. 2, pp. 152–159, Feb 2019. [Online]. Available: https://doi.org/10.1038/s41587-018-0010-1
-
[18]
ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter,
S. D. Jackman, B. P. Vandervalk, H. Mohamadi, J. Chu, S. Yeo, S. A. Hammond, G. Jahesh, H. Khan, L. Coombe, R. L. Warren, and I. Birol, “ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter,”Genome Res, vol. 27, no. 5, pp. 768–777, feb 2017
2017
-
[19]
MissForest—non-parametric missing value imputation for mixed-type data
V . C. Piro, T. H. Dadi, E. Seiler, K. Reinert, and B. Y . Renard, “ganon: precise metagenomics classification against large and up-to-date sets of reference sequences,”Bioinformatics, vol. 36, no. S1, pp. i12–i20, 07 2020. [Online]. Available: https://doi.org/10.1093/bioinformatics/ btaa458
-
[20]
A parallel algorithm for error correction in high-throughput short-read data on CUDA- enabled graphics hardware,
H. Shi, B. Schmidt, W. Liu, and W. M ¨uller-Wittig, “A parallel algorithm for error correction in high-throughput short-read data on CUDA- enabled graphics hardware,”Journal of Computational Biology, vol. 17, no. 4, pp. 603–615, 2010
2010
-
[21]
J. Chu, S. Sadeghi, A. Raymond, S. D. Jackman, K. M. Nip, R. Mar, H. Mohamadi, Y . S. Butterfield, A. G. Robertson, and I. Birol, “BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters,”Bioinformatics, vol. 30, no. 23, pp. 3402–3404, 12 2014. [Online]. Available: https://doi.org/10.1093/bioinformatics/btu558
-
[22]
Harnessing Integrated CPU-GPU System Memory for HPC: a first look into Grace Hopper,
G. Schieffer, J. Wahlgren, J. Ren, J. Faj, and I. Peng, “Harnessing Integrated CPU-GPU System Memory for HPC: a first look into Grace Hopper,” 2024. [Online]. Available: https://arxiv.org/abs/2407.07850
arXiv 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.